
|
Klik på et bogstav for at se de begreber, der er forklaringer til.
- ACE-hæmmere: Angiotensin Converting Enzyme hæmmere. ACE-hæmmere nedsætter aktiviteten af renin-angiotensin-aldosteron-systemet ved at hæmme omdannelsen af angiotensin I til II, hvorved universel vasodilatation uden sympatikusaktivering indtræder og medfører fald i blodtrykket. Anvendes typisk mod forhøjet blodtryk og hjerteinsufficiens.
- Antacida: Stoffer der neutraliserer syre produceret i mavesækken. Eller: Syreneutraliserende stoffer, der medfører neutralisering af mavesækkens pH.
- AUC: Area under the curve. Det grafiske areal under en plasmakoncentrations-tids-kurve for et lægemiddel. AUC bruges til at beskrive, hvordan kroppen eksponeres for et givent lægemiddel og anvendes til at estimere biotilgængeligheden og clearence.
- BID: Medicinsk forkortelse for bis in die = to gange dagligt.
- Biotilgængelighed, F: Den del af et oralt administreret lægemiddel, der i forhold til en intravenøs dosis når det systemiske kredsløb. Omfatter også den hastighed, hvormed dette sker. Biotilgængelighed omfatter både absorptionen over tarmvæggen (absorptionen sensu strictiori) og en evt. førstepassagemetabolisme.
- Bredspektret antibiotika: Antibiotika med virkning på et bredt spektrum af mikroorganismer, i modsætning til smalspektrede antibiotika, der kun er virksomme over for specifikke typer af mikroorganismer.
- Clearance (Cl): Forholdet mellem et lægemiddels (eller andet stofs) eliminationshastighed (mængde per tidsenhed) og dets koncentration i plasma (eller blod).
Clearance er konstant, dvs. koncentrations-uafhængig, for stoffer, der elimineres efter en 1. ordens-reaktion.
Clearance bestemmer sammen med fordelingsrummet halveringstiden. Clearance fra forskellige eliminationsorganer er additiv.
- Cmax: Den maksimale koncentration i plasma, der opnås efter lægemiddelindgift.
Ved i.v. indgift er Cmax lig Co, mens Cmax efter peroral indgift oftest først opnås efter 1-2 timer (tmax).
- CYP P450: Cytochrom-P450. Enzymsystem, som metaboliserer adskillige lægemidler via oxidering.
Oxidering udgør den kvantitativt dominerende eliminationsvej for lægemidler. CYP-enzymerne forekommer i særlig høj koncentration i leveren.
- Fald i clearance: Lægemidlet tager længere tid at få renset ud af kroppen.
- Halveringstid, t1/2: Den tid, det tager organismen (efter fordeling) at eliminere halvdelen af den tilbageværende mængde lægemiddel i kroppen.
Størrelsen er konstant og koncentrationsuafhængig for lægemidler med 1. ordens-elimination.
- Hepatisk: Vedr. leveren.
- Hypertension: Forhøjet blodtryk.
- Hypoglykæmi: Lavt blodsukker. Symptomer optræder ofte ved blodsukker lavere end 2,5 mmol/L.
- Hypotension: Lavt blodtryk.
- Hypothyreose: Nedsat funktion af skjoldbruskkirtlen som fører til nedsat dannelse af hormon (thyroxin) og dermed for lavt stofskifte.
- Inducerende lægemiddel: Når et lægemiddel forårsager øget omsætning af et andet lægemiddel via induktion af f.eks. CYP450.
- Induktion: Øget omsætning af et lægemiddel via induktion af f.eks. CYP450.
- INR: International normalized ratio. INR er en standardiseringsmetode til sammenligning af koagulationstider (protrombintider, PT). INR er således et mål for blodets evne til at koagulere.
INR har til formål at minimere forskellene mellem tromboplastinreagenser ved hjælp af en kalibreringsproces, hvor alle kommercielle tromboplastiner sammenlignes med et internationalt referencemateriale.
INR beregnes således: INR=((Patient PT)/(Middel normal PT))^ISI , og fortæller dermed hvor lang koagulationstiden er i forhold til den normale koagulationstid.
- ISI: International Sensitivity Index. Protrombintid målt med forskellige tromboplastiner kan ikke sammenlignes direkte med hinanden, f.eks. fordi sensitiviteten over for koagulationsfaktorer kan variere. For at få koagulationstider, der er så sammenlignelige som muligt, godkendte Verdenssundhedsorganisationen (WHO) i 1983 en standard reference-tromboplastin. Alle producenter af tromboplastin skal kalibrere deres reagens over for WHOs standard. Den fundne værdi betegnes International Sensitivity Index (ISI), og bruges til at beregne INR.
- Iskæmi: Ophævet eller nedsat blodforsyning af et væv i forhold til dets behov.
- Isoenzymer: Forskellige udtryksformer for et enzym. Opstår pga. af forskellige allelle gener. Eksempler ses inden for det lægemiddelomsættende system CYP450, hvor isoenzymer f.eks. er 2D6, 3A4 og 2C9.
- Kasuistik: I lægevidenskab en offentliggjort beskrivelse af et enkelt eller få sygdomstilfælde (casus (lat.): ”tilfælde, sag”).
- Lipidsænkende lægemidler: Lægemidler, der sænker visse af blodets fedtstoffer – kolesterolsænkende.
- Metabolisme: Metabolisme eller stofskifte er en generel betegnelse for den biokemiske omsætning af kemiske forbindelser i den levende organisme og dens celler. Bruges synonymt med biotransformation.
- P-gp: Permeability glycoprotein. P-gp er et cellemembran-protein, som er tilstede i epithelceller i bl.a. tarm, lever og nyrer, hvor det transporterer fremmede substanser fra blodet og ud i hhv. tarmen, galdegange og nyretubuli.
- Plasma: Plasma er den fraktion af blodet, der ikke indeholder celler. Plasma indeholder forskellige næringsstoffer, hormoner, antistoffer, koagulationsfaktorer og salte. 95% af plasma består af vand.
- PO: Per os. Via munden.
- PN medicinering: Pro re nata medicinering. Medicin, der gives efter behov.
- PT: Protrombintid. Tiden, det tager plasma at koagulere, efter tilsætning af tromboplastin (også kaldet tissue factor). Protrombintiden bruges til at vurdere blodets koagulationsevne, og anvendes især til monitorering af antikoagulationsbehandling.
- qd: Quaque die. Hver dag.
- QID: Quater in die. Fire gange dagligt.
- Renal: (af lat. renalis), vedr. nyrerne.
- Respirationsdepression: Respirationsdepression (også kaldet hypoventilation) er når frekvensen eller dybden af respirationen er utiltrækkelig til at opretholde den nødvendige gasudveksling i lungerne.
- Serotonergt syndrom: Et symptomkompleks, der skyldes overstimulering i centralnervesystemet med serotonergt aktive substanser. Symptomerne er muskelrykninger, skælven, kvalme, diarré, sved og forvirring.
- Serum: Plasma uden koagulationsfaktorer.
- SID: Semel in die. Én gang dagligt.
- SmPC: SmPC står for Summary of Product Characteristics, og er det engelske udtryk for produktresumé.
- TID: Ter in die. Tre gange dagligt.
- tmax: Det tidspunkt, hvor den maksimale plasmakoncentration af et lægemiddel indtræder. Des hurtigere absorptionshastighed, des mindre tmax.
- Total clearance: Summen af hepatisk og renal clearance. I hvilken grad disse fraktioner bidrager afhænger af, om lægemidlet primært udskilles renalt eller også undergår fase I (f.eks. via CYP) og fase II (f.eks. glukuronidering) biotransformation i leveren.
- UGT: Uridine 5'-diphospho-glucuronosyltransferase, eller UDP- glucuronosyltransferase. Glucuronyltransferaser er enzymer, som foretager konjugering (glucuronidering) af mange lægemidler og lægemiddelmetabolitter, hvorved de omdannes til stoffer, der er lettere at udskille.
- Vasodilatation: Udvidelse af kar.
- Vasokonstriktion: Sammentrækning af kar.
|
|
Formålet med Interaktionsdatabasen er at gøre behandlingen med lægemidler mere effektiv og sikker, og fremme kvaliteten i patientbehandlingen, herunder bidrage til rationel farmakoterapi. Det har været til hensigt at udvikle et redskab, der er let at anvende i den kliniske hverdag og, hvor der på højt fagligt niveau er skabt konsensus om rekommandationer og beskrivelser af interaktioner mellem lægemidler.
Interaktionsdatabasens primære evidensgrundlag er offentligt publicerede, peer-reviewed original interaktionslitteratur (kliniske studier udført på mennesker og kasuistikker) publiceret i PubMed og Embase.
Der vil således kunne forekomme uoverensstemmelse mellem andre opslagsværker, som er opbygget efter andre principper og evidenskriterier.
|
|
Etableringen af Interaktionsdatabasen var et fælles projekt mellem Danmarks Apotekerforening, Den Almindelige Danske Lægeforening, Dansk Lægemiddel Information A/S og Institut for Rationel Farmakoterapi. En projektleder og 2 farmaceuter stod for opbygningen af databasen bistået af et fagligt videnskabeligt udvalg. Desuden har der været tilknyttet eksperter indenfor forskellige fagområder. Efter en årrække under Sundhedsstyrelsen overtog Lægemiddelstyrelsen i 2015 driften og vedligeholdelsen af databasen.
|
|
Vær opmærksom på, at alle anbefalinger på Interaktionsdatabasen.dk er vejledende.
Hjemmesiden giver desuden ikke oplysninger om bivirkninger ved hvert enkelt præparat. Her henviser vi til indlægssedlen i det enkelte præparat eller til Lægemiddelstyrelsens produktresuméer.
Der kan forekomme bivirkninger, du ikke kan finde informationer om her. Dem vil vi opfordre dig til at indberette til Lægemiddelstyrelsen. Det kan du gøre på:
|
|
I denne database er lægemiddelinteraktion defineret som en ændring i enten farmakodynamikken og/eller farmakokinetikken af et lægemiddel forårsaget af samtidig behandling med et andet lægemiddel.
Interaktionsdatabasen medtager farmakodynamiske interaktioner, der ikke er umiddelbart indlysende additive (fx med forskellig virkningsmekanisme), og som kan have væsentlig klinisk betydning.
Andre faktorer, som interagerer med eller ændrer lægemiddelvirkningen så som næringsmidler (f.eks. fødemidler og kosttilskud) og nydelsesmidler (f.eks. alkohol og tobak), er ikke medtaget. Dog er medtaget lægemiddelinteraktioner med grapefrugtjuice, tranebærjuice og visse naturlægemidler.
Interaktionsdatabasens primære evidensgrundlag er offentligt publicerede, peer-reviewed original interaktionslitteratur (kliniske studier udført på mennesker samt kasuistikker) publiceret i PubMed og Embase. Desuden er interaktioner hvor data er beskrevet i produktresuméer medtaget.
I Interaktionsdatabasen findes fem forskellige symboler:
- Det røde symbol (tommelfingeren, der peger nedad) betyder, at den pågældende præparatkombination bør undgås. Denne anbefaling bliver givet i tilfælde hvor det vurderes, at den kliniske betydning er udtalt, og hvor dosisjustering ikke er mulig, eller hvis der er ligeværdige alternativer til et eller begge af de interagerende stoffer. Det røde symbol vælges også i tilfælde, hvor der vurderes at være ringe dokumenteret effekt af et eller begge stoffer, (hvor anvendelse derfor ikke findes strengt nødvendig), f.eks. for visse naturlægemidler.
- Det gule symbol (den løftede pegefinger) betyder, at kombinationen kan anvendes under visse forholdsregler. Denne anbefaling gives i tilfælde, hvor det vurderes, at den kliniske betydning er moderat til udtalt, samtidig med at den negative kliniske effekt af interaktionen kan modvirkes, enten gennem ned- eller opjustering af dosis, eller ved at forskyde indtagelsestidspunktet for det ene præparat. Anbefalingen gives også, hvis det vurderes, at kombinationen kan anvendes under forudsætning af øget opmærksomhed på effekt og/eller bivirkninger.
- Det grønne symbol (tommelfingeren, der peger opad) betyder, at kombinationen kan anvendes. Denne anbefaling gives i tilfælde, hvor det vurderes, at den kliniske betydning er uvæsentlig eller ikke tilstede.
- Det blå symbol (udråbstegnet) fremkommer i tilfælde, hvor der søges på et specifikt præparat eller en præparatkombination, som ikke findes beskrevet i Interaktionsdatabasen, men hvor der findes andre beskrevne interaktioner mellem stoffer i stofgruppen, som muligvis kan være relevante for søgningen.
- Det grå symbol (spørgsmålstegnet) fremkommer i tilfælde, hvor der er søgt på et præparat eller en præparatkombination, som (endnu) ikke er beskrevet i Interaktionsdatabasen, og hvor der heller ikke findes beskrivelser af andre præparatkombinationer mellem de to stofgrupper. En manglende beskrivelse er ensbetydende med, at Lægemiddelstyrelsen ikke har kendskab til videnskabelige undersøgelser, der undersøger en interaktion mellem den pågældende præparatkombination, og heller ikke til kasuistiske beskrivelser af en mulig interaktion. Der kan også være tale om en kombination, hvor der ikke kan drages konklusioner på baggrund af nuværende viden.
Opdatering af databasens faglige indhold foregår via litteratursøgninger som leveres via Det Kongelige Bibliotek. Litteratursøgningerne er struktureret efter veldefinerede søgekriterier og bliver løbende evalueret. Endvidere foretages yderligere håndsøgning i referencelister som kvalitetssikring af litteratursøgningerne.
Databasen bliver opdateret løbende.
Lægemiddelstyrelsens enhed Regulatorisk & Generel Medicin står for opdatering og vedligehold af Interaktionsdatabasens indhold.
Vedligehold og opdatering af databasen foretages af den faglige arbejdsgruppe, som består af 1 akademisk medarbejder og 2 studerende.
Arbejdsgruppen samarbejder med en deltidsansat speciallæge i klinisk farmakologi omkring den kliniske vurdering af lægemiddelinteraktionerne.
Interaktionsdatabasen er et opslagsværktøj, der beskriver evidensbaserede interaktioner, det vil sige interaktioner, der er dokumenteret ved publicerede kliniske studier og/eller kasuistikker. Der vil således kunne forekomme uoverensstemmelse mellem andre opslagsværker, som er opbygget efter andre principper og evidenskriterier.
Der inkluderes kun interaktioner fra offentligt publicerede, peer-reviewed original interaktionslitteratur (kliniske studier udført på mennesker samt kasuistikker) publiceret i PubMed og Embase. Desuden er interaktioner hvor data er beskrevet i produktresuméer også medtaget. Det tilstræbes at databasen opdateres snarest efter publicering, men der kan forekomme forsinkelser.
Interaktionsdatabasen beskriver interaktioner for markedsførte lægemidler, naturlægemidler samt stærke vitaminer og mineraler. I interaktionsbeskrivelserne skelnes som udgangspunkt ikke mellem forskellige dispenseringsformer. For udvalgte lægemidler skelnes dog mellem dermatologiske og systemiske formuleringer. Handelsnavnene for stærke vitaminer og mineraler, naturlægemidler samt lægemidler som ikke figurerer på medicinpriser.dk (dvs. SAD præparater) kan ikke findes på interaktionsdatabasen.
Interaktionsdatabasen omhandler ikke kosttilskud, vacciner, parenteral ernæring, elektrolytvæsker, lægemidler uden systemisk effekt og priktest (ALK).
Ja, du kan slå både lægemidler, naturlægemidler, stærke vitaminer, mineraler og enkelte frugtjuice op.
Naturlægemidler er en særlig gruppe lægemidler, der typisk indeholder tørrede planter eller plantedele, udtræk af planter eller andre naturligt forekommende bestanddele. Naturlægemidler er i lovgivningen defineret som "lægemidler, hvis indholdsstoffer udelukkende er naturligt forekommende stoffer i koncentrationer, der ikke er væsentligt større end dem, hvori de forekommer i naturen". Naturlægemidler skal godkendes af Lægemiddelstyrelsen inden de må sælges.
Stærke vitaminer og mineraler er en gruppe lægemidler, hvis indholdsstoffer udelukkende er vitaminer og/eller mineraler, og hvor indholdet af vitamin eller mineral er væsentligt højere end det normale døgnbehov hos voksne mennesker. Stærke vitaminer og mineraler kan kun godkendes til at forebygge og helbrede såkaldte mangeltilstande (og altså ikke til at behandle sygdomme). Stærke vitaminer og mineraler må kun sælges i Danmark, hvis de er godkendt af Lægemiddelstyrelsen.
Ja, du kan søge på så mange lægemidler/indholdsstoffer, du ønsker samtidig. Det gør du ved at bruge søgeboksen til højre på forsiden med overskriften ”Søg på flere præparater i kombination”. Her kan du tilføje flere felter med knappen nederst. Hvis du søger på kombinationer med mere end to slags lægemidler/indholdsstoffer, skal du være opmærksom på, at du ikke kun får ét resultat, men et antal 1+1 kombinationer. Et eksempel: Hvis du søger på samtidig brug af en p-pille, et blodtrykssænkende lægemiddel og et sovemiddel, får du 3 mulige resultater:
A: kombinationen af p-pille og blodtrykssænkende lægemiddel
B: kombinationen af p-pille og sovemiddel
C: kombinationen af blodtrykssænkende lægemiddel og sovemiddel
Du får de parvise kombinationer, der er videnskabeligt undersøgt.
Nej, du skal ikke angive dosis (500mg paracetamol) eller interval (2xdaglig), når du skal søge på et præparat eller indholdsstof. Det er kun selve præparatnavnet eller navnet på indholdsstoffet, du skal skrive. Vælg eventuelt bare navnet fra listen.
Det er desværre sådan, at der indtil videre kun kan søges på indholdsstof, når det gælder naturlægemidler.
Dette sker, når du søger på et kombinationspræparat. Når du søger på et kombinationspræparat, får du præsenteret et resultat for hvert af disse indholdsstoffer.
Indholdet i databasen er resultatet af grundige vurderinger af videnskabelige artikler og konklusioner fra humane forsøg. Hvis du kun får én interaktion på trods af, at du har indtastet flere præparater eller indholdsstoffer, skyldes det, at der endnu ikke er beskrevet (eller fundet) interaktioner af de andre indholdsstoffer i den videnskabelige litteratur.
På Lægemiddelstyrelsens hjemmeside, og i månedsbladet Rationel Farmakoterapi, juni 2015.
|
|
Lægemiddelstyrelsen
Axel Heides Gade 1
2300 København S
Tlf.nr 44 88 95 95
|
|
|
|
|
Interaktionsoplysninger
|
|
|
|
|
|
|
|
1. Præparat: Oxylan Depot - Aktivt indholdsstof: oxycodon |
|
|
 |
Interaktionsoplysninger for oxycodon og ritonavir |
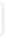 |
Hvis samtidig administration er nødvendigt, skal det overvejes at nedsætte oxycodon-dosen.
Ved samtidig administration med ritonavir (en CYP3A4-hæmmer), forventes en stigning af plasmakoncentrationen af oxycodon.
udtalt
dokumenteret
protease inhibitorer amprenavir, atazanavir, boceprevir, darunavir, fosamprenavir, indinavir, lopinavir, nelfinavir, nirmatrelvir, ombitasvir, paritaprevir, ritonavir, saquinavir, telaprevir, tipranavir opioider alfentanil, buprenorphin, codein, dextropropoxyphen, dimethylaminodiphenylbuten, fentanyl, hydromorphon, ketobemidon, levomethadon, methadon, morphin, nalbuphin, naloxon, nicomorphin, oxycodon, pentazocin, pethidin, remifentanil, sufentanil, tapentadol, tramadol Buprenorhpin Samtidig administration af buprenorphin og hhv. atazanavir og boceprevir øger koncentration af buprenorphin. Buprenophin påvirker ikke atazanavirs farmakokinetiske parametre. Tipranavir boostet med ritonavir nedsætter koncentrationen af den aktive metabolit af buprenorphin, mens der ikke blev observeret ændringer i buprenorphins farmakokinetik. Buprenorphin reducerede tipranavirs Cmax. Der ses ingen klinisk relevante farmakokinetiske ændringer ved samtidig behandling med buprenorphin og hhv. darunavir, fosamprenavir, paritaprevir, ritonavir og telaprevir. Fentanyl Samtidig administration af ritonavir og fentanyl øger koncentration af fentanyl. Methadon Samtidig administration af methadon og amprenavir kan nedsætte koncentrationen af begge stoffer. Mekanisme ukendt. Samtidig administration af methadon og hhv. boceprevir, darunavir, fosamprenavir, nelfinavir, lopinavir, ritonavir, telaprevir reducerer koncentrationen af methadon. De farmakokinetiske ændringer observeret ved co-administration af saquinavir/ritonavir og methadon vurderes, at skyldes ritonavir snarere end saquinavir. Samtidig administration af atazanavir og methadon påvirker R-methadons (aktiv) farmakokinetiske parametre, mens atazanavirs plasmakoncentration falder. Der er ikke beskrevet nogen væsentlig farmakokinetisk interaktion mellem paritaprevir og indinavir og methadon. Pethidin Samtidig administration af ritonavir og pethidin reducerer koncentration af pethidin. Samtidig administration er kontraindiceret jf. SmPC for ritonavir.
Da stofferne i de to stofgrupper er meget forskellige, er det ikke muligt at beskrive en egentlig klasseeffekt.
Litteraturgennemgang - Vis
Buprenorphin og atazanavir Ved samtidig indgift af buprenorphin (stabil dosis i mindst 2 uger) og atazanavir (400 mg) i 5 dage hos 10 methadonbrugere observeredes for buprenorphin AUC øget fra 39,5 til 76,3 ng*timer/mL (93%), Cmax øget fra 4,87 til 8,00 ng/mL (64%) og Cmin øget fra 0,84 til 1,67 ng/mL (99%). Tests afslørede ingen kognitive problemer eller abstinenssymptomer som følge af kombinationsbehandlingen. Der observeredes ingen forskel i de farmakokinetiske parametre for atazanavir sammenlignet med en kontrolgruppe (10) behandlet med atazanavir, Cance-Katz EF, Moody DE et al, 2007. I et retrospektivt kohortestudie med 303 HIV-patienter, blev sikkerheden af samtidig behandling med buprenorphin og atazanavir målt på ændringen af leverenzymerne. Der sås ingen signifikant forskel på ASAT og ALAT før og efter kombinationsbehandlingen, og der sås ingen farmakodynamiske interaktioner, Vergara-Rodriguez P, Tozzi MJ et al, 2011. Ved samtidig indgift af buprenorphin (stabil dosis i mindst 2 uger) og atazanavir/ritonavir (300/100 mg dagligt) i 5 dage til 10 methadonbrugere observeredes for buprenorphin AUC øget fra 46,2 til 77,0 ng*timer/mL (67%), Cmax øget fra 5,79 til 7,95 ng/mL (37%), og Cmin øget fra 1,09 til 1,84 ng/mL (69%). 3 deltagere rapporterede forøget sedation ved kombinationsbehandling. Ved sammenligning med en kontrolgruppe behandlet med atazanavir/ritonavir observeredes ingen forskelle i atazanavir eller ritonavirs farmakokinetiske parametre, Cance-Katz EF, Moody DE et al, 2007.
Buprenorphin og boceprevir I et interaktionsstudie med boceprevir 800mg X 3 dagligt og buprenorphin/naloxon 8/2-24/6mg dagligt er set en øgning i AUC, Cmax og Cmin for buprenorphin på 19%, 18% og 31%. Mekanismen er boceprevirs inhibering af CYP3A. SPC for Victrelis, 2013.
Buprenorphin og darunavir Ved samtidig indgift af buprenorphin/naloxen (stabil dosis i mindst 2 uger) og darunavir/ritonavir (henholdsvis 800 mg og 100 mg) i 15 dage til 11 opiod afhængige HIV-negative patienter observeres ingen signifikante farmakokinetiske ændringer for buprenorphin og darunavir. Gruber VA, Rainey PM et al, 2012. I et studium undersøgtes den eventuelle interaktion mellem opioid vedligeholdelsesterapi og darunavir/ritonavir. Raske forsøgspersoner modtog vedligeholdelsesterapi med enten metadon 55-200 mg SID (n = 16) eller buprenorfin/naloxon 16/4 mg SID (n = 5) eller 8/2 mg (n = 12) i minimum 2 uger før forsøgsstart. Herefter blev darunavir/ritonavir (600/100 mg BID) co-administreret i 7 dage. Buprenorfin/naloxon kan administreres samtidig med darunavir/ritonavir, idet AUC for buprenorfin kun faldt med 11%. Dog fandtes nor-buprenorfin eksponering at være moderat forhøjet med en stigning på 46% i AUC – den kliniske betydning heraf er uvis. Mekanisme: Buprenorfin metaboliseres via CYP3A4 og darunavir/ritonavir inhiberer CYP3A4, hvoraf ritonavir er den mest potente inhibitor, Sekar V, Tomaka F et al, 2011a.
Buprenorphin og fosamprenavir Ved samtidig indgift af buprenorphin/naloxen (stabil dosis i mindst 2 uger) og fosamprenavir/ritonavir (henholdsvis 1400 mg og 200 mg) i 15 dage til 10 opiod afhængige HIV-negative patienter observeres ingen signifikante farmakokinetiske ændringer for buprenorphin og amprenavir (den aktive metabolit af fosamprenavir). Gruber VA, Rainey PM et al, 2012.
Buprenorphin og ritonavir: Samtidig administration med 16 mg buprenorphin (hver 24. time) og 100 mg ritonavir (hver 12. time) øgede AUC og Cmax for buprenorphin med hhv. 57% og 77%. Stigninger i buprenorphins og dens aktive metabolits plasmaniveauer førte ikke til klinisk signifikante farmakodynamiske ændringer hos en gruppe af opioid-tolerante patienter.Tilpasning af buprenorphins eller ritonavirs dosis er derfor muligvis unødvendig, når disse doseres sammen. Når ritonavir bruges i kombination med en anden protease-hæmmer og buprenorphin, skal produktresuméet for den protease-hæmmer, som administreres samtidigt, ses igennem for specifik information om dosering. SmPC for Norvir, 2025 Buprenorphin og telaprevir I et interaktionsstudie med 13 hepatitis C virus-negative frivillige i stabil buprenorphin/naloxen behandling blev en mulig interaktion med telaprevir undersøgt. Personerne blev behandlet med 750mg telaprevir hver ottende time i 7 dage og for buprenorphin sås et fald i AUC og Cmax på henholdsvis 4% og 20% samt en stigning i Cmin på 6%. Luo X, Trevejo J et al, 2012, SPC for Incivo, 2013.
Buprenorphin og tipranavir I et open-label studie observeredes, ved samtidig indgift af buprenorphin/naloxon 16-24 mg/dag (stabil dosis i mindst 3 uger) og tipranavir/ritonavir (henholdsvis 500 mg og 200 mg) 2 gange dagligt i minimum 7 dage, til 10 HIV-supprimerede patienter, et 80%-fald i AUC for den aktive metabolit norbuprenorphin, men ingen effekt på farmakokinetikken af buprenorphin. Tipranavirs AUC og Cmax var 19% og 25% lavere i forhold til 161 historiske raske kontroller. Opioidtoksicitet eller abstinenser blev ikke observeret. Mekanismen er uafklaret. Bruce RD, Altice FL et al, 2009
Buprenorphin eller methadon og paritaprevir/ritonavir I et studium undersøgtes en mulig interaktion mellem 3D-regimet ombitasvir (25 mg) + paritaprevir/ritonavir (150/100 mg) og et af følgende 11 stoffer: ketoconazol (400 mg SID), warfarin (5 mg), omeprazol (40 mg SID), digoxin (0,5 mg), pravastatin (10 mg SID), rosuvastatin (5 mg SID), metadon (20-120 mg SID), buprenorphin (4-24 mg SID), naloxon (1-6 mg SID), escitalopram (10 mg) eller duloxetin (60 mg). Der fandtes hverken interaktion mellem paritaprevir/ritonavir og methadon eller paritaprevir/ritonavir og buprenorphin, Badri PS, Dutta S et al, 2016.
Buprenorphin og lopinavir: Buprenorphin: ↔
Fentanyl og lopinavir: Forøget risiko for bivirkninger (respirationsdepression, sedation) på grund af højere plasmakoncentrationer, som skyldes lopinavir/ritonavirs hæmning af CYP3A4. Omhyggelig overvågning af bivirkninger (især respirationsdepression men også sedation) anbefales, når fentanyl gives sammen med lopinavir/ritonavir. SmPC for Kaletra, 2025
Fentanyl og ritonavir Ved samtidig indgift af ritonavir (600 mg dag 1 og 900 mg dag 2) og enkeltdosis fentanyl (5 µg/kg dag 2) hos 11 raske forsøgspersoner observeredes fald i fentanyls clearance på 67%. Fentanyls AUC steg fra 4,8 til 8,8 ng/ml/timer (83%), Olkkola KT, Palkama VJ et al, 1999. Ritonavir hæmmer CYP3A4, og forventes som følge heraf at øge plasmakoncentrationerne af fentanyl. Hvis samtidig brug med nirmatrelvir/ritonavir er nødvendig, skal det overvejes at nedsætte dosis af fentanyl, samt nøje at overvåge terapeutisk virkning og bivirkninger (herunder respirationsdepression). SmPC for Paxlovid, 2025
Methadon og amprenavir Ved sammenligning af plasmakoncentrationen af methadon hos 5 misbrugspatienter før og efter 14 dages behandling med amprenavir (og abacavir) observeredes et signifikant fald på 35%. To patienter rapporterede abstinenssymptomer, Bart PA, Rizzardi PG et al, 2001. Ved indgift af amprenavir (2400 mg dagligt) hos 16 patienter i daglig behandling med methadon observeredes for R-methadon (aktiv) fald i AUC på 13%, fald i Cmax på 25% og fald i Cmin på 21%. For S-methadon (inaktiv) observeredes fald i AUC på 40%, fald i Cmax på 48% og fald i Cmin på 52%. Der rapporteredes ingen abstinenssymptomer eller behov for dosisændring af methadon, Hendrix CW, Wakeford J et al, 2004.
Methadon og atazanavir Ved indgift af atazanavir i 14 dage hos 16 patienter i methadon behandling observeres ingen statistisk signifikante ændringer i R-methadons (aktiv) kinetik, Friedland G, Andrews L et al, 2005. I et studie med 24 HIV-1-patienter var 12 patienter i behandling med både atazanavir/ritonavir 300/100 mg x 1 samt methadon oral opløsning 20-175 mg/dag, og 12 patienter var kun i behandling med atazanavir/ritonavir 300/100 mg x 1 i 4 uger. 24-timers farmakokinetiske parametre (90% CI) af atazanavir var Cmax=1714 ng/mL ved samtidig administration af methadon og 3190 ng/mL uden methadon (p=0,018); AUC=21987 ng*h/mL med methadon og 35572 ng*h/mL uden methadon (p=0,074). Ændringen i t½ for atazanavir med og uden samtidig administration af methadon var ikke signifikant (p=0,196). Mekanisme: Methadon er kendt for at være substrat uden at potensere aktiviteten af CYP2B6, -2C9, -2C19, -2D6, -3A4. Den primære metaboliske vej for atazanavir er CYP3A. Studiets resultater viser, at samtidig indtagelse af methadon nedsætter plasmakoncentration af atazanavir og ritonavir af andre ukendte grunde, Haberl A, Moesch M et al, 2010.
Methadon og boceprevir I et interaktionsstudie med methadon 20-150mg dagligt og boceprevir 800mg X3 dagligt sås for R-methadon en reduktion i AUC, Cmax og Cmin på 15%, 10% og 19%. For S-methadon sås en reduktion i AUC, Cmax og Cmin på 22%, 17% og 26%. SPC for Victrelis, 2013.
Methadon og darunavir/ritonavirI et fase 1 studium undersøgtes den eventuelle interaktion mellem opioid vedligeholdelsesterapi og darunavir/ritonavir. Raske forsøgspersoner modtog vedligeholdelsesterapi med enten metadon 55-200 mg SID (n = 16) eller buprenorfin/naloxon 16/4 mg SID (n = 5) eller 8/2 mg (n = 12) i minimum 2 uger før forsøgsstart. Herefter blev darunavir/ritonavir (600/100 mg BID) co-administreret i 7 dage. Metadon og darunavir/ritonavir co-administration af metadon og darunavir/ritonavir 600/100 mg BID resulterede i en lavere systemisk eksponering af begge metadon isomerer, hvor S-isomeren blev mere påvirket end R-isomeren med en reduktion i AUC, Cmax og Cmin på 16%, 24% og 15%. Idet R-isomeren er den biologisk aktive enantiomer, skal metadon dosis derfor højst sandsynligt ikke justeres. Mekanisme: Metadon metaboliseres via CYP3A4 og darunavir/ritonavir inhiberer CYP3A4, hvoraf ritonavir er den mest potente inhibitor. Desuden inducerer ritonavir CYP2B6, som spiller en stor rolle i udskillelsen af metadon, Sekar V, Tomaka F et al, 2011a.
Methadon og fosamprenavirVed samtidig indgift af methadon (stabil behandling) og fosamprenavir/ritonavir (1400/200 mg dagligt) i 14 dage hos 26 methadonbrugere observeredes for fald i R-methadons AUC med 18% og fald i Cmax med 21%. For S-methadon observeredes fald i AUC og Cmax med 43% for begge. Der forekom ikke symptomer på over- eller underdosering af methadon hos forsøgspersonerne, Cao YJ, Smith PF et al, 2008
Methadon og indinavir Hos 6 patienter er beskrevet uændret plasmakoncentration af methadon før og efter mindst en uges behandling med indinavir, Beauverie P, Taburet AM et al, 1998. I et overkrydsningsstudie med 12 raske forsøgspersoner behandlet med indinavir og methadon blev der ikke fundet væsentlige ændringer af de farmakokinetiske parametre for R-methadon og S-methadon og deres metabolitter. Der blev heller ikke fundet signifikante ændringer af begge enantiomerer og deres metabolitters renale clearance. Dette blev undersøgt både når methadon blev givet intravenøst eller oralt. Kharasch ED, Bedynek PS et al, 2012. Overkrydsningsstudie i 12 raske frivillige, behandlet med methadon og indinavi/ritonavir. Forsøget viste først, at indinavi/ritonavir gav en markant hæmning af hepatisk såvel som intestinal CYP3A4-aktivitet. Samtidig behandling med methadon og indinavi/ritonavir påvirkede imidlertid ikke biotilgængeligheden af methadon, hvilket tyder på, at CYP3A4 ikke har en vigtig rolle for metaboliseringen af methadon, Kharasch ED, Hoffer C et al, 2009.
Methadon og lopinavir Ved samtidig indgift af methadon og lopinavir/ritonavir til 8 HIV-smittede personer observeredes fald i AUC for methadon på 36% og fald i Cmax på 44%. Ingen patienter udviklede abstinenssymptomer, Clarke S, Mulcahy F et al, 2002. Ved tillæg af lopinavir/ritonavir (800/200 mg dagligt) til 18 HIV-smittede personer i stabil behandling med methadon (40-130 mg dagligt i mindst 4 uger) rapporteredes ingen abstinenssymptomer eller behov for dosisændring af methadon i observationsperioden (28 dage), Stevens RC, Rapaport S et al, 2003.
Methadon og nelfinavir
Ved samtidig indgift af nelfinavir og methadon hos 12 raske forsøgspersoner observeredes fald i plasmakoncentrationen af methadon på 40-50% og stigning i renal og hepatisk clearance af methadon på mellem 1,6 og 2 gange, Kharasch ED, Hoffer C et al, 2009. Hos 2 patienter er beskrevet fald i methadons plasmakoncentrationen svarende til en ratio på 0,42-0,49 ved sammenligning af før og efter mindst en uges behandling med nelfinavir, Beauverie P, Taburet AM et al, 1998. Ved samtidig indgift af nelfinavir (2500 mg dagligt) og methadon (20-140 mg dagligt, fast dosis gennem minimum 1 måned) i 8 dage hos 14 HIV-negative methadonbrugere observeredes fald i AUC for R- og S-methadon på henholdsvis 43% og 51%. Ingen patienter oplevede abstinenssymptomer eller behov for dosisjustering af methadon. Nelfinavir-koncentrationen vurderedes sammenlignelig med historiske kontroller, Hsyu PH, Lillibridge J et al, 2006a. Hos en patient i stabil behandling med methadon (100 mg dagligt gennem flere år) er beskrevet abstinenssymptomer efter tillæg af nelfinavir og behov for dosisøgning af methadon til 285 mg dagligt. Ved seponering af antiviral behandling faldt methadonbehovet til 125 mg dagligt, Cance-Katz EF, Farber S et al, 2000.
Methadon og ritonavir Ved samtidig indgift af ritonavir og methadon hos raske forsøgspersoner observeredes clearance for methadon øget med 1,5-2 gange. Renal clearance af methadon øgedes med 40-50%, Kharasch ED, Bedynek PS et al, 2008. Hos en patient er beskrevet fald i plasmakoncentrationen af methadon svarende til en ratio på 0,44 ved sammenligning af før og efter mindst en uges behandling med ritonavir, Beauverie P, Taburet AM et al, 1998.
Methadon og saquinavir Hos en patient er beskrevet uændret plasmakoncentration af methadon før og efter mindst en uges behandling med saquinavir, Beauverie P, Taburet AM et al, 1998. Ved samtidig indgift af saquinavir/ritonavir (800/800 mg dagligt i 15 dage) og enkeltdosis methadon hos 12 HIV-smittede personer observeredes for methadon fald i AUC for S-methadon på 40% og fald i R-methadons AUC på 32%. R-methadon er den mest virkningsfulde af de to isomerer. Ved korrektion for ændret proteinbinding, fandtes ikke længere statistisk signifikante ændringer i farmakokinetikken. Der optrådte ingen abstinenssymptomer eller behov for dosisændring, Gerber JG, Rosenkranz S et al, 2001. Ved samtidig indgift af saquinavir/ritonavir (1600/100 mg) og methadon i 14 dage hos 12 HIV-smittede personer observeredes ingen statistisk signifikante ændringer i farmakokinetikken for S-methadon eller R-methadon, Shelton MJ, Cloen D et al, 2004b. Hos en patient i behandling med bl.a. methadon (90 mg dagligt gennem 2 år) er beskrevet indlæggelse med abstinenssymptomer 7 dage efter skift af antiviral behandling til saquinavir/ritonavir (800/800 mg dagligt) og stavudin. Ved dosistitrering af methadon til 130 mg dagligt stabiliseredes patienten, Geletko SM og Erickson AD, 2000. Supplerende litteratur: Roche, 2012
Methadon og telaprevir I et studie med 15 personer i stabil methadonbehandling (30mg-130mg) blev telaprevirs påvirkning af methadon undersøgt. Efter 7 dags kombinations behandling med 750mg telaprevir hver ottende time i 7 dage, blev det fundet at R-methadons Cmin, Cmax og AUC blev reduceret med hhv. 31%, 29% og 29%. S-methadons Cmin, Cmax og AUC blev reduceret med hhv. 40%, 35% og 36%. Andelen af ubundet R-methadon blev ikke ændret, og der blev ikke observeret bivirkning eller ændring i kliniske parametre. Van HR, Verboven P et al, 2013.
Ritonavir og buprenorphin: ↑Buprenorphin (57 %, 77 %) Stigninger i buprenorphins og dens aktive metabolits plasmaniveauer førte ikke til klinisk signifikante farmakodynamiske ændringer hos en gruppe af opioidtolerante patienter. Tilpasning af buprenorphins dosis er derfor muligvis unødvendig, når de to doseres sammen. SmPC for Paxlovid, 2025
Ritonavir og oxycodon: ↑Oxycodon Ritonavir hæmmer CYP3A4, og forventes som følge heraf at øge plasmakoncentrationerne af oxycodon. Hvis samtidig brug med nirmatrelvir/ritonavir er nødvendig, skal det overvejes at nedsætte dosis af oxycodon, samt nøje at overvåge terapeutisk virkning og bivirkninger (herunder respirationsdepression). SmPC for Paxlovid, 2025
Nirmatrelvir/ritonavir og morphin ↓Morphin
Morphinkoncentrationerne kan blive nedsat som følge af induktion af glukuronidering forårsaget af samtidigt administreret ritonavir doseret som en farmakokinetisk forstærker. SmPC for Paxlovid, 2025
Pethidin og ritonavir Samtidig administration af pethidin og ritonavir er kontraindiceret, da der er observeret forhøjede plasmakoncentrationer af norpethidin (aktiv metabolit). Derved øges risikoen for alvorlig respirationsdepression eller andre alvorlige bivirkninger. SmPC for Ritonavir, 2021 Ved samtidig indgift af enkeltdosis petidin (50 mg) og ritonavir (1000 mg dagligt i 10 dage) hos 8 raske forsøgspersoner observeredes for petidins AUC reduceret med 67%. Norpetidins AUC (aktiv metabolit) øgedes med 47%, Piscitelli SC, Kress DR et al, 2000. Clarke S;Mulcahy F;Bergin C;Reynolds H;Boyle N;Barry M;Back DJ, Clin Infect Dis, 2002, 34:1143-1145; Absence of opioid withdrawal symptoms in patients receiving methadone and the protease inhibitor lopinavir-ritonavir A study was designed to determine the interactions, both clinical and pharmacokinetic, between methadone and lopinavir-ritonavir. Results demonstrated a 36% reduction in the methadone area under the plasma concentration-time curve after the introduction of lopinavir-ritonavir, with no coincident symptoms of opioid withdrawal and no requirement for methadone dose adjustment Haberl A;Moesch M;Nisius G;Stephan C;Bickel M;Khaykin P;Kurowski M;Brodt R;von HN, Eur J Clin Pharmacol, 2010, 66:375-381; Atazanavir plasma concentrations are impaired in HIV-1-infected adults simultaneously taking a methadone oral solution in a once-daily observed therapy setting Objective The human immundeficiency virus (HIV) protease inhibitor atazanavir is often used in once-daily observed therapy of methadone substituted former opiate drug users. We performed a matched-pairs analysis on 24 patients (12 men/women) taking atazanavir/ritonavir 300/100 mg daily plus reverse transcriptase inhibitors, with (n=12) or without (n=12) methadone co-administration. Methods Twenty-four-hour pharmacokinetic profiles of atazanavir/ritonavir were assessed at steady-state and measured by liquid chromatography-tandem mass spectrometry. The geometric mean (GM, t test) minimum and maximum plasma drug concentrations (C<sub>min</sub>, C<sub>max</sub>), area under the concentration-time curve (AUC), and total clearance (CL <sub>total</sub>) were compared between the groups of pairs, which were matched for age, sex, weight, and ethnicity. Results The GM [90% confidence interval (CI)] of the atazanavir C<sub>min</sub>, C<sub>max</sub>, and AUC of patients taking the methadone oral solution at doses of 20-175 mg/day simultaneously with antiretroviral therapy were impaired compared to patients not taking methadone oral solution: C<sub>min</sub>=315 (range 197-448) vs. 519 (279-793) ng/mL [GM ratio (GMR)=0.61, p=0.229]; C<sub>max</sub>=1714 (1238-2262) vs. 3190 (2412-4076) ng/mL (GMR=0.54, p=0.018); AUC=21,987 (15,870-29,327) vs. 35,572 (26,211-46,728) ng h/mL (GMR=0.62, p=0.074). Methadone dose, which is proportional to the amount of methadone oral solution (10 mg/mL), was significantly correlated to atazanavir C<sub>max</sub> (r<sup>2</sup>=0.40, p=0.001) and AUC (r<sup>2</sup>=0.32, p=0.006). Ritonavir pharmacokinetics was similar between the groups with C<sub>min</sub>, C<sup>max</sup>, and AUC GMR of 1.01, 0.80, and 0.96, respectively. Conclusion The partial decrease in atazanavir plasma concentrations in patients concomitantly taking racemic methadone oral solution in this daily observed therapy setting deserves further attention, and therapeutic drug monitoring should be considered. Springer-Verlag 2009 Cance-Katz EF;Farber S;Selwyn PA;O'Connor A, Am J Psychiatry, 2000, 157:481; Decrease in methodone levels with nelfinavir mesylate Geletko SM;Erickson AD, Pharmacotherapy, 2000, 20:93-94; Decreased methadone effect after ritonavir initiation Combination antiretroviral therapy including protease inhibitors such as ritonavir has added significant potency to therapy for human immunodeficiency viral (HIV) infection as well as substantial drug-drug interactions. Methadone metabolism is affected by cytochrome P450 (CYP) 3A4 inhibitors or inducers. Because ritonavir can induce CYP3A, it can decrease methadone plasma levels. An HIV-infected patient receiving methadone maintenance experienced withdrawal symptoms after ritonavir, saquinavir, and stavudine were added to his regimen; the most likely cause was ritonavir Badri PS;Dutta S;Wang H;Podsadecki TJ;Polepally AR;Khatri A;Zha J;Chiu Y;Awni WM;Menon RM, Antimicrob Agents Chemother, 2016, 60:01; Drug interactions with the direct-acting antiviral combination of ombitasvir and paritaprevir-ritonavir The two direct-acting antiviral (2D) regimen of ombitasvir and paritaprevir (administered with low-dose ritonavir) is being developed for treatment of genotype subtype 1b and genotypes 2 and 4 chronic hepatitis C virus (HCV) infection. Drug-drug interactions were evaluated in healthy volunteers to develop dosing recommendations for HCV-infected subjects. Mechanism-based interactions were evaluated for ketoconazole, pravastatin, rosuvastatin, digoxin, warfarin, and omeprazole. Interactions were also evaluated for duloxetine, escitalopram, methadone, and buprenorphine-naloxone. Ratios of geometric means with 90% confidence intervals for the maximum plasma concentration and the area under the plasma concentration-time curve were estimated to assess the magnitude of the interactions. For most medications, coadministration with the 2D regimen resulted in a <50% change in exposures. Ketoconazole, digoxin, pravastatin, and rosuvastatin exposures increased by up to 105%, 58%, 76%, and 161%, respectively, and omeprazole exposures decreased by approximately 50%. Clinically meaningful changes in ombitasvir, paritaprevir, or ritonavir exposures were not observed. In summary, all 11 medications evaluated can be coadministered with the 2D regimen, with most medications requiring no dose adjustment. Ketoconazole, digoxin, pravastatin, and rosuvastatin require lower doses, and omeprazole may require a higher dose. No dose adjustment is required for the 2D regimen Gerber JG;Rosenkranz S;Segal Y;Aberg J;D'Amico R;Mildvan D;Gulick R;Hughes V;Flexner C;Aweeka F;Hsu A;Gal J, J Acquir Immune Defic Syndr, 2001, 27:153-160; Effect of ritonavir/saquinavir on stereoselective pharmacokinetics of methadone: results of AIDS Clinical Trials Group (ACTG) 401 The effect of ritonavir 400 mg/saquinavir 400 mg twice daily on the stereoselective pharmacokinetics of methadone was examined in 12 HIV-infected, methadone-using study subjects. DESIGN: A 24-hour methadone pharmacokinetic study was performed before antiretroviral therapy was begun and after 15 days of therapy. Methadone concentration was measured by a chiral plasma assay because the drug is administered as a racemic mixture of R- and S-methadone, but only the R-isomer is active. Both changes in plasma protein binding and changes in objective and subjective opioid effect were monitored. RESULTS: Ritonavir/saquinavir administration was associated with 40% decrease in total S-methadone AUC0-24hr and 32% decrease in R-methadone area under the curve (AUC)0-24hr, and both changes were statistically significant (p =.001 for both). When AUC was corrected for the changes in protein binding induced by ritonavir/saquinavir, R-methadone free AUC0-24hr decreased 19.6% whereas the S-methadone decreased 24.6%, neither of these changes was statistically significant (p =.129 and p =.0537, respectively). This change in methadone exposure was not associated with any evidence of withdrawal from narcotics and no modification of methadone dose was required. CONCLUSIONS: Our data indicate that ritonavir/saquinavir administration is associated with induction of metabolism of methadone but this is greater for the inactive S-methadone. However, approximately 37% of the decrease in the total R-methadone exposure can be explained by protein binding displacement. Ritonavir/saquinavir can be used in HIV-infected people taking methadone without routine dose adjustments Luo X;Trevejo J;Van Heeswijk RPG;Smith F;Garga V, Antimicrob Agents Chemother, 2012, 56:3641-3647; Effect of telaprevir on the pharmacokinetics of buprenorphine in volunteers on stable buprenorphine/naloxone maintenance therapy This was an open-label, single-sequence trial in hepatitis C virus-negative volunteers on stable, individualized, buprenorphine maintenance therapy. Telaprevir at 750 mg every 8 h was coadministered with buprenorphine/naloxone (4:1 ratio as sublingual tablets) for 7 days with food. Pharmacokinetic profiles of buprenorphine, norbuprenorphine, and naloxone were measured over the 24-hour dosing interval on day -1 (buprenorphine/naloxone alone, reference) and day 7 of telaprevir coadministration (test). Geometric least-squares mean ratios and associated 90% confidence intervals of treatment ratios (test/reference) were calculated using log-transformed pharmacokinetic parameters. Opioid withdrawal symptoms were evaluated throughout the study (via questionnaires and pupillometry). Pharmacokinetic data were available for 14 and 13 volunteers on day -1 and day 7, respectively. The area under the concentration-time curve (AUC) for buprenorphine was unchanged and the maximum concentration of drug in serum (C<sub>max</sub>) for buprenorphine, C<sub>max</sub> and AUC for norbuprenorphine, and C<sub>max</sub> naxolone were modestly decreased during coadministration with telaprevir. Geometric least-squares mean ratios (90% confidence intervals) for buprenorphine were 0.80 (0.69, 0.93) for the C <sub>max</sub> and 0.96 (0.84, 1.10) for the AUC from 0 to 24 h (AUC <sub>0-24</sub>); for norbuprenorphine, values were 0.85 (0.66, 1.09) for C <sub>max</sub> and 0.91 (0.71, 1.16) for AUC<sub>0-24</sub>; for naloxone, the C<sub>max</sub> was 0.84 (0.62, 1.13). Coadministration of telaprevir did not increase withdrawal symptom frequency, and there were no serious adverse events reported during or after completion of telaprevir coadministration. Results suggest dose adjustment may not be necessary when telaprevir and buprenorphine/naloxone are coadministered. Copyright 2012, American Society for Microbiology. All Rights Reserved Vergara-Rodriguez P;Tozzi MJ;Botsko M;Nandi V;Altice F;Egan JE;O'Connor PG;Sullivan LE;Fiellin DA, J Acquir Immune Defic Syndr, 2011, 56 Suppl 1:S62-S67; Hepatic safety and lack of antiretroviral interactions with buprenorphine/naloxone in HIV-infected opioid-dependent patients BACKGROUND: The safety of buprenorphine/naloxone (bup/nx) in HIV-infected patients has not been established. Prior reports raise concern about hepatotoxicity and interactions with atazanavir. METHODS: We conducted a prospective cohort study of 303 opioid-dependent HIV-infected patients initiating bup/nx treatment. We assessed changes in aspartate aminotransferase (AST) and alanine aminotransferase (ALT) over time. We compared bup/nx doses in patients receiving the antiretroviral atazanavir to those not receiving atazanavir. We conducted surveillance for pharmacodynamic interactions. RESULTS: Median AST [37.0 vs. 37.0 units/liter (U/L) respective interquartile ranges (IQRs) 26-53 and 26-59] and ALT (33.0 vs. 33.0 U/L, respective IQRs 19-50 and 18-50) values did not change over time among 141 patients comparing pre-bup/nx exposure with post-bup/nx exposure measures. During bup/nx exposure, 207 subjects demonstrated no significant change in median AST (36.0 vs. 35.0 U/L, respective IQRs 25-57 and 25-61) and ALT (29.0 vs. 31.0 U/L, respective IQRs 19-50 and 18-50) values collected a median of 6 months apart. Analyses restricted to patients with hepatitis C and HIV co-infection yielded similar results, except a small but significant decrease in first to last AST, during treatment with bup/nx (P = 0.048). Mean bup/nx dose, ranging 16.0-17.8 mg, did not differ over time or with co-administration of atazanavir. No pharmacodynamic interactions were noted. CONCLUSIONS: Buprenorphine/naloxone did not produce measurable hepatic toxicity or pharmacodynamic interaction with atazanavir in HIV-infected opioid-dependent patients SPC for Incivo, Produktresume, 2013; Incivo (telaprevir), filmovertrukne tabletter http://www.ema.europa.eu/docs/da_DK/document_library/EPAR_-_Product_Information/human/002313/WC500115529.pdf Cance-Katz EF;Moody DE;Morse GD;Ma Q;DiFrancesco R;Friedland G;Pade P;Rainey PM, Drug Alcohol Depend, 2007, 91(2-3): 269-278-3; Interaction between buprenorphine and atazanavir or atazanavir/ritonavir Opioid addiction and HIV disease frequently co-occur. Adverse drug interactions have been reported between methadone and some HIV medications, but less is known about interactions between buprenorphine, an opioid partial agonist used to treat opioid dependence, and HIV therapeutics. This study examined drug interactions between buprenorphine and the protease inhibitors atazanavir and atazanavir/ritonavir. Opioid-dependent, buprenorphine/naloxone-maintained, HIV-negative volunteers (n = 10 per protease inhibitor) participated in two 24-h sessions to determine pharmacokinetics of (1) buprenorphine and (2) buprenorphine and atazanavir (400 mg daily) or atazanavir/ritonavir (300/100 mg daily) following administration for 5 days. Objective opiate withdrawal scale scores and mini-mental state examination were determined prior to and following antiretroviral administration to examine pharmacodynamic effects. Pharmacokinetics of atazanavir and atazanavir/ritonavir were compared in subjects and matched, healthy controls (n = 10 per protease inhibitor) to determine effects of buprenorphine. With atazanavir and atazanavir/ritonavir, respectively concentrations of buprenorphine (p < 0.001, p < 0.001), norbuprenorphine (p = 0.026, p = 0.006), buprenorphine glucuronide (p = 0.002, p < 0.001), and norbuprenorphine glucuronide (NS, p = 0.037) increased. Buprenorphine treatment did not significantly alter atazanavir or ritonavir concentrations. Three buprenorphine/naloxone-maintained participants reported increased sedation with atazanavir/ritonavir. Atazanavir or atazanavir/ritonavir may increase buprenorphine and buprenorphine metabolite concentrations and might require a decreased buprenorphine dose. copyright 2007 Elsevier Ireland Ltd. All rights reserved Gruber VA;Rainey PM;Moody DE;Morse GD;Ma Q;Prathikanti S;Pade PA;Alvanzo AA;McCance-Katz EF, Clin Infect Dis, 2012, 54:414-423; Interactions between buprenorphine and the protease inhibitors darunavir-ritonavir and fosamprenavir-ritonavir BACKGROUND: This study examined drug interactions between buprenorphine, a partial opioid agonist used for opioid dependence treatment and pain management, and the protease inhibitors (PIs) darunavir-ritonavir and fosamprenavir-ritonavir. METHODS: The pharmacokinetics of buprenorphine and its metabolites and symptoms of opioid withdrawal or excess were compared in opioid-dependent, buprenorphine-naloxone-maintained, human immunodeficiency virus (HIV)-negative volunteers (11 for darunavir-ritonavir and 10 for fosamprenavir-ritonavir) before and after 15 days of PI administration. PI pharmacokinetics and adverse effects were compared between the buprenorphine-maintained participants and an equal number of sex-, age-, race-, and weight-matched, healthy, non-opioid-dependent volunteers who received darunavir-ritonavir or fosamprenavir-ritonavir but not buprenorphine. RESULTS: There were no significant changes in buprenorphine or PI plasma levels and no significant changes in medication adverse effects or opioid withdrawal. Increased concentrations of the inactive metabolite buprenorphine-3-glucuronide suggested that darunavir-ritonavir and fosamprenavir-ritonavir induced glucuronidation of buprenorphine. CONCLUSIONS: Dose adjustments are not likely to be necessary when buprenorphine and darunavir-ritonavir or fosamprenavir-ritonavir are coadministered for the treatment of opioid dependence and HIV disease Friedland G;Andrews L;Schreibman T;Agarwala S;Daley L;Child M;Shi J;Wang Y;O'Mara E, AIDS, 2005, 19(15): 1635-1641-1641; Lack of an effect of atazanavir on steady-state pharmacokinetics of methadone in patients chronically treated for opiate addiction Background: Effective antiretroviral treatment of opiate-addicted drug users with HIV infection often requires concomitant substance abuse treatment, commonly with methadone. Pharmacological interactions between antiretroviral drugs and methadone may result in opiate withdrawal or increased side effects. Objectives: To determine if atazanavir, a once-daily protease inhibitor and moderate inhibitor of P450 CYP3A4, exhibited pharmacokinetic interactions with (R)-methadone. Methods: Methadone pharmacokinetic parameters were measured in 16 patients on chronic methadone therapy prior to and after 14 days of daily administration of atazanavir. Steady-state pharmacokinetic values for total, (R)-(active) and (S)-(inactive) isomers of methadone were derived from plasma concentrations versus time data. Symptoms of opiate withdrawal and excess were monitored. Results: For the active isomer (R)-methadone, the ratio of geometric means for coadministration with atazanavir relative to methadone alone were 1.03 [90% confidence interval (CI), 0.95-1.10] for the area under the concentration-time curve (AUC), 0.91 (90% CI, 0.84-1.00) for plasma maximal concentration and 1.11 (90% CI, 1.02-1.20) for plasma trough concentration. Confidence intervals for all three were within the no-effect or bioequivalence range of 0.80-1.25 for (R)-methadone. Inactive (S)-methadone was modestly reduced during atazanavir coadministration. Clinically relevant symptoms of opiate withdrawal or excess were not detected. Exposures to atazanavir were within range of previously reported values. Conclusions: No clinically relevant pharmacokinetic interactions were found between atazanavir and methadone. Dosage adjustment need not be recommended for either methadone or atazanavir when co-administered to patients treated for opiate abuse and HIV disease. < copyright > 2005 Lippincott Williams & Wilkins Kharasch ED;Bedynek PS;Hoffer C;Walker A;Whittington D, Anesthesiology, 2012, 116:432-447; Lack of indinavir effects on methadone disposition despite inhibition of hepatic and intestinal cytochrome P4503A (CYP3A) BACKGROUND: Methadone disposition and pharmacodynamics are highly susceptible to interactions with antiretroviral drugs. Methadone clearance and drug interactions have been attributed to cytochrome P4503A4 (CYP3A4), but actual mechanisms are unknown. Drug interactions can be clinically and mechanistically informative. This investigation assessed effects of the protease inhibitor indinavir on methadone pharmacokinetics and pharmacodynamics, hepatic and intestinal CYP3A4/5 activity (using alfentanil), and intestinal transporter activity (using fexofenadine). METHODS: Twelve healthy volunteers underwent a sequential crossover. On three consecutive days they received oral alfentanil plus fexofenadine, intravenous alfentanil, and intravenous plus oral (deuterium-labeled) methadone. This was repeated after 2 weeks of indinavir. Plasma and urine analytes were measured by mass spectrometry. Opioid effects were measured by miosis. RESULTS: Indinavir significantly inhibited hepatic and first-pass CYP3A activity. Intravenous alfentanil systemic clearance and hepatic extraction were reduced to 40-50% of control, apparent oral clearance to 30% of control, and intestinal extraction decreased by half, indicating 50% and 70% inhibition of hepatic and first-pass CYP3A activity. Indinavir increased fexofenadine area under the plasma concentration-time curve 3-fold, suggesting significant P-glycoprotein inhibition. Indinavir had no significant effects on methadone plasma concentrations, methadone N-demethylation, systemic or apparent oral clearance, renal clearance, hepatic extraction or clearance, or bioavailability. Methadone plasma concentration-effect relationships were unaffected by indinavir. CONCLUSIONS: Despite significant inhibition of hepatic and intestinal CYP3A activity, indinavir had no effect on methadone N-demethylation and clearance, suggesting little or no role for CYP3A in clinical disposition of single-dose methadone. Inhibition of gastrointestinal transporter activity had no influence of methadone bioavailability Stevens RC;Rapaport S;Maroldo-Connelly L;Patterson JB;Bertz R, J Acquir Immune Defic Syndr, 2003, 33:650-651; Lack of methadone dose alterations or withdrawal symptoms during therapy with lopinavir/ritonavir Kharasch ED;Bedynek PS;Park S;Whittington D;Walker A;Hoffer C, Clin Pharmacol Ther, 2008, 84:497-505; Mechanism of ritonavir changes in methadone pharmacokinetics and pharmacodynamics: I. Evidence against CYP3A mediation of methadone clearance Ritonavir diminishes methadone plasma concentrations, an effect attributed to CYP3A induction, but the actual mechanisms are unknown. We determined ritonavir effects on stereoselective methadone pharmacokinetics and clinical effects (pupillary miosis) in healthy human immunodeficiency virus-negative volunteers. Subjects received intravenous plus oral (deuterium-labeled) racemic methadone after no ritonavir, short-term (3-day) ritonavir, and steady-state ritonavir. Acute and steady-state ritonavir, respectively, caused 1.5- and 2-fold induction of systemic and apparent oral R- and S-methadone clearances. Ritonavir increased renal clearance 40-50%, and stereoselectively (S > R) increased hepatic methadone N-demethylation 50-80%, extraction twofold, and clearance twofold. Bioavailability was unchanged despite significant inhibition of intestinal P-glycoprotein. Intestinal and hepatic CYP3A was inhibited > 70%. Ritonavir shifted methadone plasma concentration-miosis curves leftward and upward. Rapid ritonavir induction of methadone clearance results from increased renal clearance and induced hepatic metabolism. Induction of methadone metabolism occurred despite profound CYP3A inhibition, suggesting no role for CYP3A in clinical methadone metabolism and clearance. Ritonavir may alter methadone pharmacodynamics Bart PA;Rizzardi PG;Gallant S;Golay KP;Baumann P;Pantaleo G;Eap CB, Ther Drug Monit, 2001, 23:553-555; Methadone blood concentrations are decreased by the administration of abacavir plus amprenavir Abacavir and amprenavir, a nucleoside reverse transcription inhibitor and a protease inhibitor, respectively, are new drugs used for the treatment of HIV. Methadone blood concentrations were measured in five addict patients receiving methadone maintenance therapy before and after introduction of abacavir plus amprenavir. The administration of these two drugs for a median period of 14 days resulted in a significant reduction (P = 0.043) of methadone concentration, with a median decrease to 35% of the original concentration (range 28-87%). Two patients reported on several occasions nausea in the morning before the intake of the daily methadone dose, which is compatible with withdrawal reaction to opioids. Because amprenavir is a cytochrome P4503A4 substrate and is involved in the metabolism of methadone, reduction of methadone concentrations could be explained by an induction of cytochrome P4503A4 Kharasch ED;Hoffer C;Whittington D;Walker A;Bedynek PS, Anesthesiology, 2009, 110:660-672; Methadone pharmacokinetics are independent of cytochrome P4503A (CYP3A) activity and gastrointestinal drug transport: Insights from methadone interactions with ritonavir/indinavir BACKGROUND: Methadone clearance is highly variable, and drug interactions are problematic. Both have been attributed to CYP3A, but actual mechanisms are unknown. Drug interactions can provide such mechanistic information. Ritonavir/indinavir, one of the earliest protease inhibitor combinations, may inhibit CYP3A. We assessed ritonavir/indinavir effects on methadone pharmacokinetics and pharmacodynamics, intestinal and hepatic CYP3A activity, and intestinal transporters (P-glycoprotein) activity. CYP3A and transporters were assessed with alfentanil and fexofenadine, respectively. METHODS: Twelve healthy human immunodeficiency virus-negative volunteers underwent a sequential three-part crossover. On three consecutive days, they received oral alfentanil/fexofenadine, intravenous alfentanil, and intravenous plus oral (deuterium-labeled) methadone, repeated after acute (3 days) and steady-state (2 weeks) ritonavir/indinavir. Plasma and urine analytes were measured by mass spectrometry. Opioid effects were assessed by miosis. RESULTS: Alfentanil apparent oral clearance was inhibited more than 97% by both acute and steady-state ritonavir/indinavir, and systemic clearance was inhibited more than 90% due to diminished hepatic and intestinal extraction. Ritonavir/indinavir increased fexofenadine area under the plasma concentration-time curve four-to five-fold, suggesting significant inhibition of gastrointestinal P-glycoprotein. Ritonavir/indinavir slightly increased methadone N-demethylation, but it had no significant effects on methadone plasma concentrations or on systemic or apparent oral clearance, renal clearance, hepatic extraction or clearance, or bioavailability. Ritonavir/indinavir had no significant effects on methadone plasma concentration-effect relationships. CONCLUSIONS: Inhibition of both hepatic and intestinal CYP3A activity is responsible for ritonavir/indinavir drug interactions. Methadone disposition was unchanged, despite profound inhibition of CYP3A activity, suggesting little or no role for CYP3A in clinical methadone metabolism and clearance. Methadone bioavailability was unchanged, despite inhibition of gastrointestinal P-glycoprotein activity, suggesting that this transporter does not limit methadone intestinal absorption Van HR;Verboven P;Vandevoorde A;Vinck P;Snoeys J;Boogaerts G;De PE;Van Solingen-Ristea R;Witek J;Garg V, Antimicrob Agents Chemother, 2013, 57:May; Pharmacokinetic interaction between telaprevir and methadone Hepatitis C virus (HCV) antibody is present in most patients enrolled in methadone maintenance programs. Therefore, interactions between the HCV protease inhibitor telaprevir and methadone were investigated. The pharmacokinetics of R- and S-methadone were measured after administration of methadone alone and after 7 days of telaprevir (750 mg every 8 h [q8h]) coadministration in HCV-negative subjects on stable, individualized methadone therapy. Unbound R-methadone was measured in predose plasma samples before and during telaprevir coadministration. Safety and symptoms of opioid withdrawal were evaluated throughout the study. In total, 18 subjects were enrolle 2 discontinued prior to receiving telaprevir. The minimum plasma concentration in the dosing interval (C<sub>min</sub>), the maximum plasma concentration (C<sub>max</sub>), and the area under the plasma concentration- time curve fromh 0 (time of administration) to 24 h postdose (AUC<sub>0-24</sub>) for R-methadone were reduced by 31%, 29%, and 29%, respectively, in the presence of telaprevir. The AUC<sub>0-24</sub> ratio of S-methadone/R-methadone was not altered. The median unbound percentage of R-methadone increased by 26% in the presence of telaprevir. The R-methadone median (absolute) unbound C<sub>min</sub> values in the absence (10.63 ng/ml) and presence (10.45 ng/ml) of telaprevir were similar. There were no symptoms of opioid withdrawal and no discontinuations due to adverse events. In summary, exposure to total R-methadone was reduced by approximately 30% in the presence of telaprevir, while the exposure to unbound R-methadone was unchanged. No symptoms of opioid withdrawal were observed. These results suggest that dose adjustment of methadone is not required when initiating telaprevir treatment. (This study has been registered at ClinicalTrials.gov under registration no. NCT00933283.) Copyright 2013, American Society for Microbiology. All Rights Reserved Hsyu PH;Lillibridge J;Daniels E;Kerr BM, Biopharm Drug Dispos, 2006, a, 27(2): 61-68-68; Pharmacokinetic interaction of nelfinavir and methadone in intravenous drug users The effect of nelfinavir 1250 mg twice daily (b.i.d.) on the pharmacokinetics of methadone was determined in 14 HIV-negative methadone users. Design: The methadone dose (20-140 mg/day) was stabilized and fixed for at least 1 month before nelfinavir (1250 mg b.i.d. for 8 days) was added to the regimen. The concentrations of methadone enantiomers were measured before and during nelfinavir treatment, and the concentrations of nelfinavir and its active metabolite, AG1402, were measured during nelfinavir treatment. Adverse events and withdrawal/intoxication symptoms were monitored throughout the study. Results: Nelfinavir reduced the area under the concentration-time curve of R-methadone, and S-methadone by 43% and 51%, respectively. Nelfinavir and AG1402 concentrations were within the normal range of historical data, and no subject experienced withdrawal symptoms during the study or required dose adjustment during or after the study. Conclusions: Although nelfinavir reduced the plasma concentrations of both R- and S-methadone, it seems to have no impact on the maintenance dose of methadone. A routine reduction of methadone dose is not recommended when coadministered with nelfinavir. Copyright < copyright > 2005 John Wiley & Sons, Ltd Bruce RD;Altice FL;Moody DE;Lin S;Fang WB;Sabo JP;Wruck JM;Piliero PJ;Conner C;Andrews L;Friedland GH, Drug and Alcohol Dependence, 2009, 105:234-239; Pharmacokinetic interactions between buprenorphine/naloxone and tipranavir/ritonavir in HIV-negative subjects chronically receiving buprenorphine/naloxone HIV-infected patients with opioid dependence often require opioid replacement therapy. Pharmacokinetic interactions between HIV therapy and opioid dependence treatment medications can occur. HIV-seronegative subjects stabilized on at least 3 weeks of buprenorphine/naloxone (BUP/NLX) therapy sequentially underwent baseline and steady-state pharmacokinetic evaluation of open-label, twice daily tipranavir 500 mg co-administered with ritonavir 200 mg (TPV/r). Twelve subjects were enrolled and 10 completed the study. Prior to starting TPV/r, the geometric mean BUP AUC0-24 h and Cmax were 43.9 ng h/mL and 5.61 ng/mL, respectively. After achieving steady-state with TPV/r ([greater-than or equal to]7 days), these values were similar at 43.7 ng h/mL and 4.84 ng/mL, respectively. Similar analyses for norBUP, the primary metabolite of BUP, demonstrated a reduction in geometric mean for AUC0-24 h [68.7-14.7 ng h/mL; ratio = 0.21 (90% CI 0.19-0.25)] and Cmax [4.75-0.94 ng/mL; ratio = 0.20 (90% CI 0.17-0.23)]. The last measurable NLX concentration (Clast) in the concentration-time profile, never measured in previous BUP/NLX interaction studies with antiretroviral medications, was decreased by 20%. Despite these pharmacokinetic effects on BUP metabolites and NLX, no clinical opioid withdrawal symptoms were noted. TPV steady-state AUC0-12 h and Cmax decreased 19% and 25%, respectively, and Cmin was relatively unchanged when compared to historical control subjects receiving TPV/r alone. No dosage modification of BUP/NLX is required when co-administered with TPV/r. Though mechanistically unclear, it is likely that decreased plasma RTV levels while on BUP/NLX contributed substantially to the decrease in TPV levels. BUP/NLX and TPV/r should therefore be used cautiously to avoid decreased efficacy of TPV in patients taking these agents concomitantly. copyright 2009 Elsevier Ireland Ltd. All rights reserved Sekar V;Tomaka F;Lefebvre E;De PM;Vangeneugden T;van den Brink W;Hoetelmans R, Tomt indhold, 2011, a, 51:271-278; Pharmacokinetic interactions between darunavir/ritonavir and opioid maintenance therapy using methadone or buprenorphine/naloxone Hendrix CW;Wakeford J;Wire MB;Lou Y;Bigelow GE;Martinez E;Christopher J;Fuchs EJ;Snidow JW, Pharmacotherapy, 2004, 24:1110-1121; Pharmacokinetics and pharmacodynamics of methadone enantiomers after coadministration with amprenavir in opioid-dependent subjects STUDY OBJECTIVE: To investigate the steady-state pharmacokinetics of methadone enantiomers when coadministered with amprenavir. DESIGN: Prospective, open-label, within-subject pharmacokinetic study. SETTING: University research center. SUBJECTS: Nineteen opioid-dependent, methadone-maintained, healthy individuals were enrolled. INTERVENTION: On study day 1, subjects received their usual once-daily dose of methadone alone. On study days 2-11, they received the same once-daily methadone dose plus amprenavir 1200 mg twice/day. Serial blood samples were collected over 24 hours on study days 1 and 11 for measurement of plasma R- and S-methadone, and over 12 hours on day 11 for serum amprenavir concentrations. MEASUREMENTS AND MAIN RESULTS: Standard pharmacokinetic parameters were determined from the concentrations and compared between the two treatments (methadone alone vs methadone with amprenavir). Subjects served as their own control for methadone comparisons, and amprenavir comparisons were made by using a historic control group (38 healthy men). Opioid-effect measures were assessed throughout the study. Coadministration of amprenavir with methadone resulted in a 3-4-hour delay in plasma R- and S-methadone enantiomer peak concentrations at steady state (Cmax-ss). The active R-methadone enantiomer area under the plasma concentration-time curve during a dosing interval (AUCt-ss, Cmax-ss, and the minimum plasma concentration at steady state (Cmin-ss) were decreased by 13%, 25%, and 21%, respectively, after coadministration of methadone and amprenavir. The inactive S-enantiomer AUCt-ss, Cmax-ss, and Cmin-ss were decreased by 40%, 48%, and 52%, respectively. No clinically significant changes were noted in opioid pharmacodynamic effects, and there was no evidence of opioid withdrawal. No methadone dosage was changed in any subject. CONCLUSION: No a priori adjustment in methadone dosage is required during coadministration with amprenavir as there is only a small effect on R-methadone exposure and no evidence of opioid withdrawal Cao YJ;Smith PF;Wire MB;Lou Y;Lancaster CT;Causon RC;Bigelow GE;Martinez E;Fuchs EJ;Radebaugh C;McCabe S;Hendrix CW, Pharmacotherapy, 2008, 28:863-874; Pharmacokinetics and pharmacodynamics of methadone enantiomers after coadministration with fosamprenavir-ritonavir in opioid-dependent subjects STUDY OBJECTIVE: To compare steady-state pharmacokinetics and pharmacodynamics of methadone enantiomers when coadministered with fosamprenavir 700 mg-ritonavir 100 mg twice/day. DESIGN: Open-label, single-sequence, two-period crossover, drug-interaction study. SETTING: Two university-affiliated research centers. SUBJECTS: Twenty-six opioid-dependent, methadone-maintained, healthy adults. INTERVENTION: Subjects received their usual daily dose of methadone alone for 4 days (period 1). Subjects then received the same daily dose of methadone plus fosamprenavir 700 mg-ritonavir 100 mg twice/day for 14 days (period 2). MEASUREMENTS AND MAIN RESULTS: Blood was collected on days 1-4 (period 1) and on days 11-14 (period 2) for plasma R- and S-methadone concentrations; amprenavir concentrations were assessed during period 2. Opioid-effect measures were assessed in each study period. Subjects served as their own controls for comparison of period 1 with period 2. Coadministration of fosamprenavir-ritonavir with methadone reduced plasma total R-methadone area under the plasma concentration-time curve over the dosing interval at steady state (AUC tau-ss) by 18%, maximum concentration at steady state (Cmax-ss) by 21%, and concentration at the end of the dosing interval at steady state (Ctau-ss) by 11%; time to reach Cmax-ss (Tmax) was delayed by 1.75 hours. Coadministration of fosamprenavir-ritonavir with methadone also reduced plasma total S-methadone AUC tau-ss and Cmax-ss by 43% each, Ctau-ss by 41%, and delayed Tmax by 0.85 hours. Fosamprenavir-ritonavir administered with methadone did not alter plasma amprenavir pharmacokinetics compared with historical control data; nor did it alter the unbound R-methadone at 2 and 6 hours after methadone dosing. Pharmacodynamic indexes remained essentially unchanged after adding fosamprenavir-ritonavir to methadone. No subject demonstrated opioid intoxication or withdrawal, or requested methadone dosage modification. CONCLUSION: No adjustment in the dosages of either methadone or fosamprenavir 700 mg-ritonavir 100 mg twice/day is required during coadministration, on the basis of the small reduction in total R-methadone exposure, no change in unbound R-methadone, no clinically important opioid effects, and no change in amprenavir exposure SPC for Prezista, Tomt indhold, 2014; Prezista (darunavir), 100mg/ml oral suspension Olkkola KT;Palkama VJ;Neuvonen PJ, Anesthesiology, 1999, 91:681-685; Ritonavir's role in reducing fentanyl clearance and prolonging its half-life BACKGROUND: The human immunodeficiency virus protease inhibitor ritonavir is a potent inhibitor of the cytochrome P450 3A4 enzyme, and ritonavir's concomitant administration with the substrates of this enzyme may lead to dangerous drug interactions. METHODS: The authors investigated possible interactions between ritonavir and intravenously administered fentanyl in a double-blind, placebo-controlled, cross-over study in two phases. Twelve healthy volunteers received orally ritonavir or placebo for 3 days; the dose of ritonavir was 200 mg three times on the first day and 300 mg three times on the second. The last dose of ritonavir 300 mg or placebo was given on the morning of the third day. On the second day, 2 h after the afternoon pretreatment dose, fentanyl 5 microg/kg was injected intravenously in 2 min with naloxone to moderate its effects, and 15 timed venous blood samples were collected for 18 h. RESULTS: Ritonavir reduced the clearance of fentanyl by 67% from 15.6+/-8.2 to 5.2+/-2.0 ml x min(-1) x kg(-1) (P<0.01). The area under the fentanyl plasma concentration-time curve from 0 to 18 h was increased from 4.8+/-2.7 to 8.8+/-2.3 ng x ml(-1) x h(-1) by ritonavir (P<0.01). Ritonavir did not affect the initial concentrations and the steady-state volume of distribution of fentanyl. One subject discontinued participation before fentanyl administration because of severe side effects, and during the study 8 of the remaining 11 subjects reported nausea. CONCLUSIONS: Ritonavir can inhibit the metabolism of fentanyl significantly, so caution should be exercised if fentanyl is given to patients receiving ritonavir medication SmPC for Kaletra, Produktresume, 2025; SmPC for Kaletra https://www.ema.europa.eu/da/documents/product-information/kaletra-epar-product-information_da.pdf SmPC for Norvir, Produktresume, 2025; SmPC for Norvir https://www.ema.europa.eu/da/documents/product-information/norvir-epar-product-information_da.pdf SmPC for Paxlovid, Produktresume, 2025; SmPC for Paxlovid https://www.ema.europa.eu/da/documents/product-information/paxlovid-epar-product-information_da.pdf Roche, Tomt indhold, 2012; Summary of product characteristics: Invirase 200 mg hard capsules Piscitelli SC;Kress DR;Bertz RJ;Pau A;Davey R, Pharmacotherapy, 2000, 20:549-553; The effect of ritonavir on the pharmacokinetics of meperidine and normeperidine STUDY OBJECTIVE: To determine the effects of ritonavir on the pharmacokinetics of meperidine and normeperidine. DESIGN: Open-label, crossover, pharmacokinetic study. SETTING: United States government research hospital. SUBJECTS: Eight healthy volunteers who tested negative for the human immunodeficiency virus. INTERVENTION: Subjects received oral meperidine 50 mg and had serial blood samples collected for 48 hours. They then received ritonavir 500 mg twice/day for 10 days, followed by administration of a second 50-mg meperidine dose and collection of serial samples. MEASUREMENTS AND MAIN RESULTS: Plasma samples were assayed for meperidine, normeperidine, and ritonavir. Meperidine's area under the curve (AUC) decreased in all subjects by a mean of 67+/-4% in the presence of ritonavir (p<0.005). Mean +/- SD maximum concentration was decreased from 126+/-47 to 51+/-21 ng/ml. Normeperidine's mean AUC was increased 47%, suggesting induction of hepatic metabolism. CONCLUSION: Meperidine's AUC is significantly reduced, not increased, by concomitant ritonavir. Based on these findings, the risk of narcotic-related adverse effects from this combination appears to be minimal. However, increased concentrations of normeperidine suggest a potential for toxicity with increased dosages or long-term therapy Shelton MJ;Cloen D;DiFrancesco R;Berenson CS;Esch A;de Caprariis PJ;Palic B;Schur JL;Bugge CJ;Ljungqvist A;Espinosa O;Hewitt RG, J Clin Pharmacol, 2004, b, 44:293-304; The effects of once-daily saquinavir/minidose ritonavir on the pharmacokinetics of methadone Twelve methadone-maintained HIV-negative subjects were given saquinavir/ritonavir (SQV/rtv) 1600 mg/100 mg once daily for 14 days. Pharmacokinetic evaluations of total and unbound methadone enantiomers (R and S) were conducted before and after SQV/rtv. SQV/rtv was well tolerated, with no ACTG Grade 3-4 adverse events, no evidence of sedation, and no changes in methadone dose. For R-methadone (active isomer), C(max), AUC(0-24 h), and C(min) were unchanged, but percent unbound 4 hours after dosing was reduced by 12%. For S-methadone, no differences in pharmacokinetic parameters of total drug were seen, but unbound concentrations were reduced by 15% and 21% at 4 and 24 hours after dosing, respectively. SQV trough concentrations exceeded the anticipated EC(50) (50 ng/mL) in 10/12 subjects, persisting for at least 6 hours after the final dose in 4/6 subjects. Once-daily SQV/rtv in methadone-maintained subjects is safe and not associated with any clinically significant interaction with methadone during 14 days of concomitant administration Beauverie P;Taburet AM;Dessalles MC;Furlan V;Touzeau D, AIDS, 1998, 12:2510-2511; Therapeutic drug monitoring of methadone in HIV-infected patients receiving protease inhibitors SmPC for Ritonavir, Produktresume, 2021; Viatris ApS https://www.ema.europa.eu/en/documents/product-information/ritonavir-mylan-epar-product-information_da.pdf SPC for Victrelis, Produktresume, 2013; Victrelis (boceprevir), 200 mg hårde kapsler
|
|
|
|
|
|
|