
|
Klik på et bogstav for at se de begreber, der er forklaringer til.
- ACE-hæmmere: Angiotensin Converting Enzyme hæmmere. ACE-hæmmere nedsætter aktiviteten af renin-angiotensin-aldosteron-systemet ved at hæmme omdannelsen af angiotensin I til II, hvorved universel vasodilatation uden sympatikusaktivering indtræder og medfører fald i blodtrykket. Anvendes typisk mod forhøjet blodtryk og hjerteinsufficiens.
- Antacida: Stoffer der neutraliserer syre produceret i mavesækken. Eller: Syreneutraliserende stoffer, der medfører neutralisering af mavesækkens pH.
- AUC: Area under the curve. Det grafiske areal under en plasmakoncentrations-tids-kurve for et lægemiddel. AUC bruges til at beskrive, hvordan kroppen eksponeres for et givent lægemiddel og anvendes til at estimere biotilgængeligheden og clearence.
- BID: Medicinsk forkortelse for bis in die = to gange dagligt.
- Biotilgængelighed, F: Den del af et oralt administreret lægemiddel, der i forhold til en intravenøs dosis når det systemiske kredsløb. Omfatter også den hastighed, hvormed dette sker. Biotilgængelighed omfatter både absorptionen over tarmvæggen (absorptionen sensu strictiori) og en evt. førstepassagemetabolisme.
- Bredspektret antibiotika: Antibiotika med virkning på et bredt spektrum af mikroorganismer, i modsætning til smalspektrede antibiotika, der kun er virksomme over for specifikke typer af mikroorganismer.
- Clearance (Cl): Forholdet mellem et lægemiddels (eller andet stofs) eliminationshastighed (mængde per tidsenhed) og dets koncentration i plasma (eller blod).
Clearance er konstant, dvs. koncentrations-uafhængig, for stoffer, der elimineres efter en 1. ordens-reaktion.
Clearance bestemmer sammen med fordelingsrummet halveringstiden. Clearance fra forskellige eliminationsorganer er additiv.
- Cmax: Den maksimale koncentration i plasma, der opnås efter lægemiddelindgift.
Ved i.v. indgift er Cmax lig Co, mens Cmax efter peroral indgift oftest først opnås efter 1-2 timer (tmax).
- CYP P450: Cytochrom-P450. Enzymsystem, som metaboliserer adskillige lægemidler via oxidering.
Oxidering udgør den kvantitativt dominerende eliminationsvej for lægemidler. CYP-enzymerne forekommer i særlig høj koncentration i leveren.
- Fald i clearance: Lægemidlet tager længere tid at få renset ud af kroppen.
- Halveringstid, t1/2: Den tid, det tager organismen (efter fordeling) at eliminere halvdelen af den tilbageværende mængde lægemiddel i kroppen.
Størrelsen er konstant og koncentrationsuafhængig for lægemidler med 1. ordens-elimination.
- Hepatisk: Vedr. leveren.
- Hypertension: Forhøjet blodtryk.
- Hypoglykæmi: Lavt blodsukker. Symptomer optræder ofte ved blodsukker lavere end 2,5 mmol/L.
- Hypotension: Lavt blodtryk.
- Hypothyreose: Nedsat funktion af skjoldbruskkirtlen som fører til nedsat dannelse af hormon (thyroxin) og dermed for lavt stofskifte.
- Inducerende lægemiddel: Når et lægemiddel forårsager øget omsætning af et andet lægemiddel via induktion af f.eks. CYP450.
- Induktion: Øget omsætning af et lægemiddel via induktion af f.eks. CYP450.
- INR: International normalized ratio. INR er en standardiseringsmetode til sammenligning af koagulationstider (protrombintider, PT). INR er således et mål for blodets evne til at koagulere.
INR har til formål at minimere forskellene mellem tromboplastinreagenser ved hjælp af en kalibreringsproces, hvor alle kommercielle tromboplastiner sammenlignes med et internationalt referencemateriale.
INR beregnes således: INR=((Patient PT)/(Middel normal PT))^ISI , og fortæller dermed hvor lang koagulationstiden er i forhold til den normale koagulationstid.
- ISI: International Sensitivity Index. Protrombintid målt med forskellige tromboplastiner kan ikke sammenlignes direkte med hinanden, f.eks. fordi sensitiviteten over for koagulationsfaktorer kan variere. For at få koagulationstider, der er så sammenlignelige som muligt, godkendte Verdenssundhedsorganisationen (WHO) i 1983 en standard reference-tromboplastin. Alle producenter af tromboplastin skal kalibrere deres reagens over for WHOs standard. Den fundne værdi betegnes International Sensitivity Index (ISI), og bruges til at beregne INR.
- Iskæmi: Ophævet eller nedsat blodforsyning af et væv i forhold til dets behov.
- Isoenzymer: Forskellige udtryksformer for et enzym. Opstår pga. af forskellige allelle gener. Eksempler ses inden for det lægemiddelomsættende system CYP450, hvor isoenzymer f.eks. er 2D6, 3A4 og 2C9.
- Kasuistik: I lægevidenskab en offentliggjort beskrivelse af et enkelt eller få sygdomstilfælde (casus (lat.): ”tilfælde, sag”).
- Lipidsænkende lægemidler: Lægemidler, der sænker visse af blodets fedtstoffer – kolesterolsænkende.
- Metabolisme: Metabolisme eller stofskifte er en generel betegnelse for den biokemiske omsætning af kemiske forbindelser i den levende organisme og dens celler. Bruges synonymt med biotransformation.
- P-gp: Permeability glycoprotein. P-gp er et cellemembran-protein, som er tilstede i epithelceller i bl.a. tarm, lever og nyrer, hvor det transporterer fremmede substanser fra blodet og ud i hhv. tarmen, galdegange og nyretubuli.
- Plasma: Plasma er den fraktion af blodet, der ikke indeholder celler. Plasma indeholder forskellige næringsstoffer, hormoner, antistoffer, koagulationsfaktorer og salte. 95% af plasma består af vand.
- PO: Per os. Via munden.
- PN medicinering: Pro re nata medicinering. Medicin, der gives efter behov.
- PT: Protrombintid. Tiden, det tager plasma at koagulere, efter tilsætning af tromboplastin (også kaldet tissue factor). Protrombintiden bruges til at vurdere blodets koagulationsevne, og anvendes især til monitorering af antikoagulationsbehandling.
- qd: Quaque die. Hver dag.
- QID: Quater in die. Fire gange dagligt.
- Renal: (af lat. renalis), vedr. nyrerne.
- Respirationsdepression: Respirationsdepression (også kaldet hypoventilation) er når frekvensen eller dybden af respirationen er utiltrækkelig til at opretholde den nødvendige gasudveksling i lungerne.
- Serotonergt syndrom: Et symptomkompleks, der skyldes overstimulering i centralnervesystemet med serotonergt aktive substanser. Symptomerne er muskelrykninger, skælven, kvalme, diarré, sved og forvirring.
- Serum: Plasma uden koagulationsfaktorer.
- SID: Semel in die. Én gang dagligt.
- SmPC: SmPC står for Summary of Product Characteristics, og er det engelske udtryk for produktresumé.
- TID: Ter in die. Tre gange dagligt.
- tmax: Det tidspunkt, hvor den maksimale plasmakoncentration af et lægemiddel indtræder. Des hurtigere absorptionshastighed, des mindre tmax.
- Total clearance: Summen af hepatisk og renal clearance. I hvilken grad disse fraktioner bidrager afhænger af, om lægemidlet primært udskilles renalt eller også undergår fase I (f.eks. via CYP) og fase II (f.eks. glukuronidering) biotransformation i leveren.
- UGT: Uridine 5'-diphospho-glucuronosyltransferase, eller UDP- glucuronosyltransferase. Glucuronyltransferaser er enzymer, som foretager konjugering (glucuronidering) af mange lægemidler og lægemiddelmetabolitter, hvorved de omdannes til stoffer, der er lettere at udskille.
- Vasodilatation: Udvidelse af kar.
- Vasokonstriktion: Sammentrækning af kar.
|
|
Formålet med Interaktionsdatabasen er at gøre behandlingen med lægemidler mere effektiv og sikker, og fremme kvaliteten i patientbehandlingen, herunder bidrage til rationel farmakoterapi. Det har været til hensigt at udvikle et redskab, der er let at anvende i den kliniske hverdag og, hvor der på højt fagligt niveau er skabt konsensus om rekommandationer og beskrivelser af interaktioner mellem lægemidler.
Interaktionsdatabasens primære evidensgrundlag er offentligt publicerede, peer-reviewed original interaktionslitteratur (kliniske studier udført på mennesker og kasuistikker) publiceret i PubMed og Embase.
Der vil således kunne forekomme uoverensstemmelse mellem andre opslagsværker, som er opbygget efter andre principper og evidenskriterier.
|
|
Etableringen af Interaktionsdatabasen var et fælles projekt mellem Danmarks Apotekerforening, Den Almindelige Danske Lægeforening, Dansk Lægemiddel Information A/S og Institut for Rationel Farmakoterapi. En projektleder og 2 farmaceuter stod for opbygningen af databasen bistået af et fagligt videnskabeligt udvalg. Desuden har der været tilknyttet eksperter indenfor forskellige fagområder. Efter en årrække under Sundhedsstyrelsen overtog Lægemiddelstyrelsen i 2015 driften og vedligeholdelsen af databasen.
|
|
Vær opmærksom på, at alle anbefalinger på Interaktionsdatabasen.dk er vejledende.
Hjemmesiden giver desuden ikke oplysninger om bivirkninger ved hvert enkelt præparat. Her henviser vi til indlægssedlen i det enkelte præparat eller til Lægemiddelstyrelsens produktresuméer.
Der kan forekomme bivirkninger, du ikke kan finde informationer om her. Dem vil vi opfordre dig til at indberette til Lægemiddelstyrelsen. Det kan du gøre på:
|
|
I denne database er lægemiddelinteraktion defineret som en ændring i enten farmakodynamikken og/eller farmakokinetikken af et lægemiddel forårsaget af samtidig behandling med et andet lægemiddel.
Interaktionsdatabasen medtager farmakodynamiske interaktioner, der ikke er umiddelbart indlysende additive (fx med forskellig virkningsmekanisme), og som kan have væsentlig klinisk betydning.
Andre faktorer, som interagerer med eller ændrer lægemiddelvirkningen så som næringsmidler (f.eks. fødemidler og kosttilskud) og nydelsesmidler (f.eks. alkohol og tobak), er ikke medtaget. Dog er medtaget lægemiddelinteraktioner med grapefrugtjuice, tranebærjuice og visse naturlægemidler.
Interaktionsdatabasens primære evidensgrundlag er offentligt publicerede, peer-reviewed original interaktionslitteratur (kliniske studier udført på mennesker samt kasuistikker) publiceret i PubMed og Embase. Desuden er interaktioner hvor data er beskrevet i produktresuméer medtaget.
I Interaktionsdatabasen findes fem forskellige symboler:
- Det røde symbol (tommelfingeren, der peger nedad) betyder, at den pågældende præparatkombination bør undgås. Denne anbefaling bliver givet i tilfælde hvor det vurderes, at den kliniske betydning er udtalt, og hvor dosisjustering ikke er mulig, eller hvis der er ligeværdige alternativer til et eller begge af de interagerende stoffer. Det røde symbol vælges også i tilfælde, hvor der vurderes at være ringe dokumenteret effekt af et eller begge stoffer, (hvor anvendelse derfor ikke findes strengt nødvendig), f.eks. for visse naturlægemidler.
- Det gule symbol (den løftede pegefinger) betyder, at kombinationen kan anvendes under visse forholdsregler. Denne anbefaling gives i tilfælde, hvor det vurderes, at den kliniske betydning er moderat til udtalt, samtidig med at den negative kliniske effekt af interaktionen kan modvirkes, enten gennem ned- eller opjustering af dosis, eller ved at forskyde indtagelsestidspunktet for det ene præparat. Anbefalingen gives også, hvis det vurderes, at kombinationen kan anvendes under forudsætning af øget opmærksomhed på effekt og/eller bivirkninger.
- Det grønne symbol (tommelfingeren, der peger opad) betyder, at kombinationen kan anvendes. Denne anbefaling gives i tilfælde, hvor det vurderes, at den kliniske betydning er uvæsentlig eller ikke tilstede.
- Det blå symbol (udråbstegnet) fremkommer i tilfælde, hvor der søges på et specifikt præparat eller en præparatkombination, som ikke findes beskrevet i Interaktionsdatabasen, men hvor der findes andre beskrevne interaktioner mellem stoffer i stofgruppen, som muligvis kan være relevante for søgningen.
- Det grå symbol (spørgsmålstegnet) fremkommer i tilfælde, hvor der er søgt på et præparat eller en præparatkombination, som (endnu) ikke er beskrevet i Interaktionsdatabasen, og hvor der heller ikke findes beskrivelser af andre præparatkombinationer mellem de to stofgrupper. En manglende beskrivelse er ensbetydende med, at Lægemiddelstyrelsen ikke har kendskab til videnskabelige undersøgelser, der undersøger en interaktion mellem den pågældende præparatkombination, og heller ikke til kasuistiske beskrivelser af en mulig interaktion. Der kan også være tale om en kombination, hvor der ikke kan drages konklusioner på baggrund af nuværende viden.
Opdatering af databasens faglige indhold foregår via litteratursøgninger som leveres via Det Kongelige Bibliotek. Litteratursøgningerne er struktureret efter veldefinerede søgekriterier og bliver løbende evalueret. Endvidere foretages yderligere håndsøgning i referencelister som kvalitetssikring af litteratursøgningerne.
Databasen bliver opdateret løbende.
Lægemiddelstyrelsens enhed Regulatorisk & Generel Medicin står for opdatering og vedligehold af Interaktionsdatabasens indhold.
Vedligehold og opdatering af databasen foretages af den faglige arbejdsgruppe, som består af 1 akademisk medarbejder og 2 studerende.
Arbejdsgruppen samarbejder med en deltidsansat speciallæge i klinisk farmakologi omkring den kliniske vurdering af lægemiddelinteraktionerne.
Interaktionsdatabasen er et opslagsværktøj, der beskriver evidensbaserede interaktioner, det vil sige interaktioner, der er dokumenteret ved publicerede kliniske studier og/eller kasuistikker. Der vil således kunne forekomme uoverensstemmelse mellem andre opslagsværker, som er opbygget efter andre principper og evidenskriterier.
Der inkluderes kun interaktioner fra offentligt publicerede, peer-reviewed original interaktionslitteratur (kliniske studier udført på mennesker samt kasuistikker) publiceret i PubMed og Embase. Desuden er interaktioner hvor data er beskrevet i produktresuméer også medtaget. Det tilstræbes at databasen opdateres snarest efter publicering, men der kan forekomme forsinkelser.
Interaktionsdatabasen beskriver interaktioner for markedsførte lægemidler, naturlægemidler samt stærke vitaminer og mineraler. I interaktionsbeskrivelserne skelnes som udgangspunkt ikke mellem forskellige dispenseringsformer. For udvalgte lægemidler skelnes dog mellem dermatologiske og systemiske formuleringer. Handelsnavnene for stærke vitaminer og mineraler, naturlægemidler samt lægemidler som ikke figurerer på medicinpriser.dk (dvs. SAD præparater) kan ikke findes på interaktionsdatabasen.
Interaktionsdatabasen omhandler ikke kosttilskud, vacciner, parenteral ernæring, elektrolytvæsker, lægemidler uden systemisk effekt og priktest (ALK).
Ja, du kan slå både lægemidler, naturlægemidler, stærke vitaminer, mineraler og enkelte frugtjuice op.
Naturlægemidler er en særlig gruppe lægemidler, der typisk indeholder tørrede planter eller plantedele, udtræk af planter eller andre naturligt forekommende bestanddele. Naturlægemidler er i lovgivningen defineret som "lægemidler, hvis indholdsstoffer udelukkende er naturligt forekommende stoffer i koncentrationer, der ikke er væsentligt større end dem, hvori de forekommer i naturen". Naturlægemidler skal godkendes af Lægemiddelstyrelsen inden de må sælges.
Stærke vitaminer og mineraler er en gruppe lægemidler, hvis indholdsstoffer udelukkende er vitaminer og/eller mineraler, og hvor indholdet af vitamin eller mineral er væsentligt højere end det normale døgnbehov hos voksne mennesker. Stærke vitaminer og mineraler kan kun godkendes til at forebygge og helbrede såkaldte mangeltilstande (og altså ikke til at behandle sygdomme). Stærke vitaminer og mineraler må kun sælges i Danmark, hvis de er godkendt af Lægemiddelstyrelsen.
Ja, du kan søge på så mange lægemidler/indholdsstoffer, du ønsker samtidig. Det gør du ved at bruge søgeboksen til højre på forsiden med overskriften ”Søg på flere præparater i kombination”. Her kan du tilføje flere felter med knappen nederst. Hvis du søger på kombinationer med mere end to slags lægemidler/indholdsstoffer, skal du være opmærksom på, at du ikke kun får ét resultat, men et antal 1+1 kombinationer. Et eksempel: Hvis du søger på samtidig brug af en p-pille, et blodtrykssænkende lægemiddel og et sovemiddel, får du 3 mulige resultater:
A: kombinationen af p-pille og blodtrykssænkende lægemiddel
B: kombinationen af p-pille og sovemiddel
C: kombinationen af blodtrykssænkende lægemiddel og sovemiddel
Du får de parvise kombinationer, der er videnskabeligt undersøgt.
Nej, du skal ikke angive dosis (500mg paracetamol) eller interval (2xdaglig), når du skal søge på et præparat eller indholdsstof. Det er kun selve præparatnavnet eller navnet på indholdsstoffet, du skal skrive. Vælg eventuelt bare navnet fra listen.
Det er desværre sådan, at der indtil videre kun kan søges på indholdsstof, når det gælder naturlægemidler.
Dette sker, når du søger på et kombinationspræparat. Når du søger på et kombinationspræparat, får du præsenteret et resultat for hvert af disse indholdsstoffer.
Indholdet i databasen er resultatet af grundige vurderinger af videnskabelige artikler og konklusioner fra humane forsøg. Hvis du kun får én interaktion på trods af, at du har indtastet flere præparater eller indholdsstoffer, skyldes det, at der endnu ikke er beskrevet (eller fundet) interaktioner af de andre indholdsstoffer i den videnskabelige litteratur.
På Lægemiddelstyrelsens hjemmeside, og i månedsbladet Rationel Farmakoterapi, juni 2015.
|
|
Lægemiddelstyrelsen
Axel Heides Gade 1
2300 København S
Tlf.nr 44 88 95 95
|
|
|
|
|
Interaktionsoplysninger
|
|
|
|
|
|
|
|
1. Indholdsstof simvastatin |
|
|
 |
|
|
2. Indholdsstof amlodipin |
|
|
|
|
|
3. Indholdsstof acetylsalicylsyre |
|
|
|
|
|
4. Indholdsstof paracetamol |
|
|
|
|
|
6. Indholdsstof furosemid |
|
|
|
|
|
7. Indholdsstof bendroflumethiazid |
|
|
|
|
|
8. Præparat: Losartan "Medical Valley" - Aktivt indholdsstof: losartan |
|
|
|
|
|
9. Indholdsstof enalapril |
|
|
|
|
|
10. Indholdsstof atorvastatin |
|
|
|
|
|
11. Indholdsstof ibuprofen |
|
|
|
|
|
12. Præparat: Citalopram "1A Farma" - Aktivt indholdsstof: citalopram |
|
|
|
|
|
13. Indholdsstof metoprolol |
|
|
|
|
|
14. Pantoprazol "Actavis" - (ingen præparater og/eller indholdsstoffer matchede dette søgeord!) |
|
|
|
|
|
15. Indholdsstof omeprazol |
|
|
|
|
|
16. Indholdsstof metformin |
|
|
|
|
|
17. Præparat: Lansoprazol "Medical Valley" - Aktivt indholdsstof: lansoprazol |
|
|
|
|
|
18. Oroxine - (ingen præparater og/eller indholdsstoffer matchede dette søgeord!) |
|
|
 |
Interaktionsoplysninger for amlodipin og simvastatin |
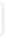 |
Øget opmærksomhed på bivirkninger som myopati og rhabdomyolyse, særligt ved høj dosis af simvastatin.
Forskydning af doseringstidspunkt for de to stoffer på 4 timer kan dog mindske stigningen i AUC for simvastatin. Produktresumeer for amlodipin og simvastatin anbefaler, at dosis af simvastatin begrænses til 20 mg dagligt. Som alternativ kan pravastatin og rosuvastatin overvejes, da de ikke metaboliseres via CYP3A4, jf. klasseeffekt To studier har vist, at amlodipin kan øge eksponeringen for simvastatin (Cmax og AUC steg i et af studierne med faktor 1,4 og 1,3 - uden at det dog førte til en ændring af den kolesterolsænkende effekt af simvastatin). Mekanisme: amlodipin hæmmer simvastatins metabolisme via CYP3A4.
moderat
veldokumenteret
calciumantagonister amlodipin, diltiazem, felodipin, isradipin, lacidipin, lercanidipin, nifedipin, nimodipin, nitrendipin, verapamil Hmg coa reduktase-inhibitorer atorvastatin, cerivastatin, fluvastatin, lovastatin, pravastatin, rosuvastatin, simvastatinStatiner, med undtagelse af pravastatin og rosuvastatin, omsættes overvejende via CYP3A4. Blandt calciumantagonisterne betragtes verapamil og diltiazem som værende moderate CYP3A4 inhibitorer, og amlodipin har også en CYP3A4 inhiberende effekt. Ved kombination ses derfor typisk øgede koncentrationer (AUC) af statiner (i mindre grad rosuvastatin og pravastatin), og flere kasuistikker rapporterer om tilfælde af rhabdomyolyse ved kombination mellem de CYP3A4-inhiberende calciumantagonister og simvastatin og atorvastatin. Atorvastatin og amlodipine er godkendt som kombinationsprodukt i bl.a. USA (Caduet©) og der er ikke set farmakokinetiske interaktioner for denne kombination.
Nogle studier har fundet ændring i eksponeringen for calciumantagonister: Atorvastatin og lovastatin kan således forårsage en stigning i eksponering for verapamil (hhv. 42,8% og 62,8% øget AUC for verapamil).
Der er i litteraturen ikke fundet prospektive undersøgelser eller kasuistikker omhandlende interaktion mellem statiner og felopidin, nimodipin og nitredipin.
Litteraturgennemgang - Vis
Atorvastatin og diltiazem Hos to patienter (Gladding P, Pilmore H et al, 2004;Lewin JJ, Nappi JM et al, 2002b) observeres myopati ved kombinationsbehandling med 40 mg atorvastatin og 120 mg diltiazem. Der er dog ikke målt serumkoncentrationer af atorvastatin i de to cases, og ætiologien for myopatien er derfor uafklaret. Atorvastatin og verapamil 12 raske forsøgspersoner fik verapamil (po., 60 mg) samtidigt med behandling med atorvastatin (po., 40 mg), Choi DH, Shin WG et al, 2008. Resultatet var en signifikant ændring af verapamils farmakokinetik og blandt blev AUC øget med 42,8%. Der blev ikke målt koncentrationer af atorvastatin i dette studium. Den kliniske betydning er ikke undersøgt, men der sås ikke ændring af blodtryk eller puls i det beskrevne studium.
Atorvastatin og amlodipne
Cmax og AUC blev undersøgt for kombinationen af atorvastatin og amlopidine i et 2-vejs crossover RCT blandt 126 raske forsøgspersoner (dosis kombinationer 80mg/10mg til 10mg/5mg). Cmax og AUC for begge stoffer svarede til værdierne fra produktresumeerne for de stoffer alene Chung M, Calcagni A et al, 2006 Studiet blev udført i forlængelse af FDA’s godkendelsen af Atorvastatin/Amplodipin kombinationsproduktet Caduet© SPS for CADUET, 2015. Efficacy, safety og farmakodynamisk interatkion mellem amlodine og atorvastatin blev undersøgt i et RCT blandt 1660 forsøgspersoner med forhøjet blodtryk og forhøjet kolesterolniveau Preston RA, Harvey P et al, 2007a. Produkterne blev indgivet samtidigt med forskellige dosis kombinationer (5mg/10mg – 10mg/80mg) og sammenlignet med produkterne indgivet alene. Samlet set påvirkede samtidig administration ikke den kolesterolsænkende effekt af atorvastatin, eller den blodtryksnedsættende effekt af amlodipin. Dog sås en øget LDL reduktion ved kombinationen af atorvastatin 10 mg og 5 mg amlodipine sammenliget med atorvastatin 10 mg alene.
Fluvastatin og amlodipin Coadministration af amlodipin og fluvastatin resulterede ikke i signifikante ændringer i AUC, Cmax eller Clearance for fluvastatin, sammenlignet med personer hvor fluvastatin blev givet alene. Ligeledes sås ingen ændringer i AUC, Cmax eller Clerance for amlodipin når forsøgsperonerne fik amlodipin givet alene, Prasad PP, Stypinski D et al, 2004b. Fluvastatin og lercanidipin I et studie fik 8 raske kvinder på skift og med min. 7 dages "washout" enten 40 mg fluvastatin, 20 mg lercanidipin eller begge lægemidler samtidigt, Boralli VB, Coelho EB et al, 2009. AUC for Fluvastatin blev let øget ved sammentidig indgift i lercanidipin. Samtidig faldt AUC for lercanidipin let ved samtidig indgift af fluvastatin. Den kliniske betydning af denne interaktion er uafklaret, men er formodentlig ringe. Lovastatin og diltiazem Ved samtidig indgift af en enkelt dosis lovastatin og steady-state koncentrationer af diltiazem hos 10 raske forsøgspersoner (Azie NE, Brater DC et al, 1998) sås en stigning i AUC for lovastatin med en faktor 3,5. Halveringstiden blev ikke signifikant påvirket. Mekanisme: øget biotilgængelighed på grund af hæmning af P-glycoprotein og CYP3A4 i tarmen. I et randomiseret 2-vejs overkrydsningsstudie modtog 10 raske frivillige forsøgspersoner hhv. lovastatin (20 mg) og lovastain (20 mg) 1 time efter intravenøs infusion af diltiazem. Blodprøver blev opsamlet i op til 25 timer efter administration og viste ingen signifikant påvirkning af AUC, Cmax, t½ og tmax for lovastatin. Interaktion mellem lovastatin og diltiazem formodes således at være en ”first pass” effekt der vil vise sig ved skift fra IV til oral administration, Masica AL, Azie NE et al, 2000. Lovastatin og isradipin Ved indgift af lovastatin og isradipin hos 12 raske forsøgspersoner (Zhou LX, Finley DK et al, 1995) observeres et fald i AUC for lovastatin på ca. 40%, hos de seks mandlige forsøgspersoner, men intet fald hos kvinder. Lovastatin og verapamil 14 raske forsøgspersoner modtog, i et standard 2x2 overkrydsningsstudie af Choi D, Chung J et al, 2010, 60 mg verapamil p.o. med (n=7) eller uden (n=7) 20 mg lovastatin p.o. Verapamil AUC steg 62,8% (p<0,01), Cmax steg 32,1% (p<0,05) og den relative biotilgængelighed øgedes 1,77 gange. Farmakokinetik af metabolitten, norverapamil ændredes ikke signifikant. Mekanisme: Lovastatin inhiberer p-glycoprotein og CYP3A4 i tarm/lever og påvirker dermed omsætningen af verapamil.
Pravastatin og diltiazem Der er ud fra et farmakokinetisk synspunkt ikke fundet nogen interaktion mellem pravastatin og diltiazem, Azie NE, Brater DC et al, 1998. Ved gennemgang af amerikanske bivirkningsindberetninger til FDA findes ingen øget risiko for forekomst af rhabdomyolyse ved kombination af pravastatin og CYP3A4 inhibitorer, herunder diltiazem, Rowan C, Brinker AD et al, 2009.
Pravastatin og verapamil Ved gennemgang af amerikanske bivirkningsindberetninger til FDA findes ingen øget risiko for rhabdomyolyse ved kombination af pravastatin og CYP3A4 inhibitorer, herunder verapamil, Rowan C, Brinker AD et al, 2009. Simvastatin og amlodipin I et studie med 8 patienter med hyperkolesterolæmi og hypertension, blev amlodipin og simvastatin (begge 5 mg od) givet samtidigt i 4 uger, Nishio S, Watanabe H et al, 2005. Resultatet var en øgning af Cmax og AUC for simvastatin med hhv. en faktor 1,4 og 1,3 - uden at det dog førte til en ændring af den kolesterolsænkende effekt af simvastatin. En forskydning af doseringstidspunkt for de to stoffer på 4 timer kan dog mindske stigningen i AUC for simvastatin Park CG, Lee H et al, 2010a I et koreansk randomiseret open-label cross-over studie på 48 raske unge mænd blev simvastatin (40 mg) givet alene eller samtidig med amlodipin (10 mg) dagligt i 9 dage, efterfulgt af 14 dages udvaskningsperiode. Kombinationsbehandling medførte en stigning i AUC og Cmax på 1,8 og 1,9 gange for simvastatin og 1,9 og 2,3 gange for simvastatinsyre. Forfatterne udviklede udfra forsøgsdata en model til at forudsige optimal dosisreduktion af simvastatin og kom frem til at anbefale en dosisreduktion på 40% ved kombinationsbehandling med amlodipin, Son H, Lee D et al, 2014. Simvastatin og diltiazem Ved samtidig indgift af enkelt dosis simvastatin og steady-state koncentrationer af diltiazem hos 10 raske forsøgspersoner (Mousa O, Brater DC et al, 2000) sås en stigning i AUC for simvastatin med en faktor 5. Halveringstiden blev forlænget med en faktor 2,3. Watanabe H, Kosuge K et al, 2004 finder at AUC og Cmax for simvastatin fordobles efter 4 ugers samtidig behandling med 5 mg simvastatin og 90mg. diltiazem dgl. Hos to patienter (Kanathur N, Mathai MG et al, 2001a) er der observeret rhabdomyolyse efter 3 måneders kombinationsbehandling med simvastatin og diltiazem. Dertil er der hos to patienter (Gladding P, Pilmore H et al, 2004) efter kort tids kombinationsbehandling med simvastatin konstateret udvikling af rhabdomyolyse og efterfølgende nyresvigt. Hos en af patienterne medførte dette endvidere døden. Der er dog ikke målt serumkoncentrationer af simvastatin i disse cases, og det er således uafklaret om der har været en klinisk betydende interaktion til grund for disse forløb. Et retrospektivt registerstudie af 135 hypertensionspatienter i behandling med forskellige calciumkanalblokkere, som var blevet opstartet i simvastatin behandling, undersøgte om en interaktion mellem diltiazem og simvastatin forøger kolesterolfaldet hos patienter, der er i behandling med både simvastatin og diltiazem. Kolesterolfaldet for 19 patienter på diltiazem (gennemsnitsdosis på 226 mg dgl) var 33% sammenlignet med 24,7% for de resterende 116 patienter (gennemsnitsforskel 8,6%, 95% CI 1,1-12,2%, P<0,02), Yeo KR, Yeo WW et al, 1999. Ved gennemgang af amerikanske bivirkningsindberetninger til FDA, finder forfatterne evidens for øget risiko for rhabdomyolyse ved samtidig behandling med simvastatin (CYP3A4-substrat) og en række CYP3A4 inhibitorer, herunder diltiazem, Rowan C, Brinker AD et al, 2009. Mekanisme: Diltiazem hæmmer CYP3A4, som omsætter simvastatin, hvilket resulterer i en ophobning af simvastatin og dermed et øget kolesterolfald. Interaktionen kunne anvendes klinisk til opnåelse et anbefalet kolestreolfald under anvendelse af lavere doser af simvastatin og samtidig diltiazem behandling. Simvastatin og lacidipin Ved indgift af simvastatin og lacidipin hos 18 raske forsøgspersoner (Ziviani L, Da Ros L et al, 2001) observeres ændringer i både simvastatin og den aktive metabolit simvastatin syre`s kinetik. AUC for simvastatin steg ca. 25%, mens halveringstiden forblev uændret. For simvastatin syre sås ligeledes stigninger i AUC på ca. 35%. Disse ændringer vurderes dog ikke at være klinisk betydningsfulde. Simvastatin og lercanidipin Ved gentagen administrering af 20 mg lercanidipin sammen med 40 mg simvastatin blev AUC for lercanidipin ikke ændret signifikant, medens AUC for simvastatin blev forøget med 56 % og dets aktive metabolit β-hydroxysyre med 28 %. Ændringerne vurderes ikke at være klinisk relevante, SPC for Zanidip, 2007. Simvastatin og verapamil Ved samtidig indgift af simvastatin og verapamil hos 12 raske forsøgspersoner (Kantola T, Kivisto KT et al, 1998b), sås stigning i AUC for den aktive simvastatin syre med en faktor 2,8. Halveringstiden blev ikke bestemt. Den kliniske betydning af denne interaktion er uafklaret. Ved gennemgang af amerikanske bivirkningsindberetninger til FDA blev fundet evidens for øget risiko for rhabdomyolyse ved samtidig behandling med simvastatin (CYP3A4-substrat) og en række CYP3A4 inhibitorer, herunder verapamil, Rowan C, Brinker AD et al, 2009. Supplerende litteratur: Peces R og Pobes A, 2001a; SPC for simvastatin 16572 Son H;Lee D;Lim LA;Jang SB;Roh H;Park K, Drug Metabolism and Pharmacokinetics, 2014, 29:2014; Development of a pharmacokinetic interaction model for co-administration of simvastatin and amlodipine A model for drug interaction between amlodipine and simvastatin was developed using concentration data obtained from a multiple-dose study consisting of single- and co-administration of amlodipine and simvastatin conducted in healthy Koreans. Amlodipine concentrations were assumed to influence the clearance of simvastatin and simvastatin acid, which as well as the oral bioavailability was allowed to vary depending on genetic polymorphisms of metabolic enzymes. Covariate effects on drug concentrations were also considered. The developed model yielded a 46% increase in simvastatin bioavailability and a 13% decrease in simvastatin clearance when amlodipine 10 mg was co-administered. When CYP3A4/5 polymorphisms were assessed by a mixture model, extensive metabolizers yielded a decrease in simvastatin bioavailability of 81% and a decrease in simvastatin clearance by 4.6 times as compared to poor metabolizers. Sixty percent of the usual dose was the optimal simvastatin dose that can minimize the interaction with amlodipine 10 mg. Age and weight had significant effects on amlodipine concentrations. In conclusion, this study has quantitatively described the pharmacokinetic interaction between simvastatin and amlodipine using a modeling approach. Given that the two drugs are often prescribed together, the developed model is expected to contribute to more efficient and safer drug treatment when they are co-administered. Copyright © 2014 by the Japanese Society for the Study of Xenobiotics (JSSX) Choi DH;Shin WG;Choi JS, Eur J Clin Pharmacol, 2008, 64(5):445-449; Drug interaction between oral atorvastatin and verapamil in healthy subjects: Effects of atorvastatin on the pharmacokinetics of verapamil and norverapamil Aim: It has been reported that verapamil and atorvastatin are inhibitors of both P-glycoprotein (P-gp) and microsomal cytochrome P450 (CYP) 3A4, and verapamil is a substrate of both P-gp and CYP3A4. Thus, it could be expected that atorvastatin would alter the absorption and metabolism of verapamil. Methods: The pharmacokinetic parameters of verapamil and one of its metabolites, norverapamil, were compared after oral administration of verapamil (60 mg) in the presence or absence of oral atorvastatin (40 mg) in 12 healthy volunteers. Results: Pharmacokinetics of verapamil were significantly altered by the coadministration of atorvastatin compared with those of without atorvastatin. For example, the total area under the plasma-concentration time curve to the last measured time, 24 h, in plasma (AUC(0-24) (h)) of verapamil increased significantly by 42.8%. Thus, the relative bioavailability increased by the same magnitude with atorvastatin. Although the AUC (0-24) (h) of norverapamil was not significantly different between two groups of humans, the AUC(0-24) (h, norverapamil)/ AUC(0-24) (h, verapamil) ratio was significantly reduced (27.5% decrease) with atorvastatin. Conclusion: The above data suggest that atorvastatin could inhibit the absorption of verapamil via inhibition of P-gp and/or the metabolism of verapamil by CYP3A4 in humans. copyright 2008 Springer-Verlag Boralli VB;Coelho EB;Sampaio SA;Marques MP;Lanchote VL, J Clin Pharmacol, 2009, 49:205-211; Enantioselectivity in the pharmacokinetic interaction between fluvastatin and lercanidipine in healthy volunteers Hypertension and dyslipidemia are independent risk factors for cardiovascular mortality and are frequently present in the same patient. Fluvastatin (FV), used to reduce cholesterol levels, and lercanidipine (LER), used to control blood pressure, are marketed as racemic mixtures. Therapeutic activities are 30-fold higher for (+ )-3R,5S-FV and 100- to 200-fold higher for S-LER compared with their respective antipodes. The present study describes the enantioselective pharmacokinetic interaction between LER and FV in healthy volunteers. A crossover randomized study was conducted in 3 phases on 8 volunteers treated with a single oral racemic dose of LER (20 mg) or FV (40 mg) or LER plus FV. Serial blood samples were collected from 0 to 24 hours. Plasma concentrations of the LER and FV enantiomers were determined by liquid chromatography/tandem mass spectrometry, and pharmacokinetic parameters were evaluated using the WinNonlin software. The Wilcoxon and Mann-Whitney tests (P <.05) were used to analyze enantiomer ratios and the pharmacokinetic drug interaction. Data are expressed as medians. In monotherapy, the kinetic disposition of both FV and LER was enantioselective. AUC values were significantly higher for (-)-3S,5R-FV than for (+)-3R,5S-FV (358.20 vs 279.68 ng.h/mL) and for S-LER compared with R-LER (13.90 vs 11.88 ng.h/mL). The pharmacokinetic parameters of FV were not enantioselective when combined with LER (AUC: (-)-3S,5R-FV: 325.21; (+)-3R,5S-FV: 316.44 ng.h/mL). There was a significant reduction in S-LER (8.06 vs 13.90 ng.h/mL) and R-LER (6.76 vs 11.88 ng.h/mL) AUC values when FV was coadministered. In conclusion, the interaction between FV-LER might be clinically relevant because AUC values of (+)-3R,5S-FV were increased when LER was coadministered, and AUC values of the 2 LER enantiomers were reduced when FV was coadministered. copyright 2009 the American College of Clinical Pharmacology Yeo KR;Yeo WW;Wallis EJ;Ramsay LE, Br J Clin Pharmacol, 1999, 48:610-615; Enhanced cholesterol reduction by simvastatin in diltiazem-treated patients AIMS: To investigate whether an interaction between diltiazem and the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor simvastatin may enhance the cholesterol-lowering response to simvastatin in diltiazem-treated patients. METHODS: One hundred and thirty-five patients attending the Sheffield hypertension clinic who started consecutively on simvastatin for primary or secondary prevention of coronary heart disease (CHD) during the 2 years June, 1996-May 1998 were surveyed. From the clinic records we extracted and recorded absolute and percentage cholesterol responses to the starting dose of simvastatin and coprescription of diltiazem. RESULTS: The cholesterol reduction for the 19 patients on diltiazem was 33.3% compared with 24.7% in the remaining 116 patients (median difference 8.6%, 95% CI 1.1-12.2%, P<0.02). The interindividual variability of cholesterol response to simvastatin was greater for patients not taking diltiazem than for those patients taking diltiazem. The ratio of the variances in response for the nondiltiazem group relative to the diltiazem group was 1.34 at 10 mg simvastatin daily (not significant, 95% CI 0.16-4.11), and 3.42 at 20 mg daily (P<0.01, 95% CI 1.26-7.18). Concurrent diltiazem therapy (P<0.04), age (P=0.001) and starting dose of simvastatin (P=0.002) were found to be significant independent predictors of percentage cholesterol response. CONCLUSIONS: Patients who take both simvastatin and diltiazem may need lower doses of simvastatin to achieve the recommended reduction in cholesterol. The pharmacokinetic and pharmacodynamic aspects of this interaction need further study to confirm an enhanced effect on cholesterol reduction, and exclude an increased risk of adverse events Kantola T;Kivisto KT;Neuvonen PJ, Clin Pharmacol Ther, 1998, b, 64:177-182; Erythromycin and verapamil considerably increase serum simvastatin and simvastatin acid concentrations OBJECTIVE: To study the effects of erythromycin and verapamil on the pharmacokinetics of simvastatin, an inhibitor of 3-hydroxy-3- methylglutaryl coenzyme A reductase. METHODS: A randomized, double- blind crossover study was performed with three phases separated by a washout period of 3 weeks. Twelve young, healthy volunteers took orally either 1.5 gm/day erythromycin, 240 mg/day verapamil, or placebo for 2 days. On day 2, 40 mg simvastatin was administered orally. Serum concentrations of simvastatin, simvastatin acid, erythromycin, verapamil, and norverapamil were measured for up to 24 hours. RESULTS: Erythromycin and verapamil increased mean peak serum concentration (Cmax) of unchanged simvastatin 3.4-fold (p < 0.001) and 2.6-fold (p < 0.05) and the area under the serum simvastatin concentration-time curve from time zero to 24 hours [AUC(0-24)] 6.2-fold (p < 0.001) and 4.6- fold (p < 0.01). Erythromycin increased the mean Cmax of active simvastatin acid fivefold (p < 0.001) and the AUC(0-24) 3.9-fold (p < 0.001). Verapamil increased the Cmax of simvastatin acid 3.4-fold (p < 0.001) and the AUC(0-24) 2.8-fold (p < 0.001). There was more than tenfold interindividual variability in the extent of simvastatin interaction with both erythromycin and verapamil. CONCLUSIONS: Both erythromycin and verapamil interact considerably with simvastatin, probably by inhibiting its cytochrome P450 (CYP) 3A4-mediated metabolism. Concomitant administration of erythromycin, verapamil, or other potent inhibitors of CYP3A4 with simvastatin should be avoided. As an alternative, the dosage of simvastatin should be reduced considerably, that is, by about 50% to 80%, at least when a simvastatin dosage higher than 20 mg/day is used. Possible adverse effects, such as elevation of creatine kinase level and muscle tenderness, should be closely monitored when such combinations are used Nishio S;Watanabe H;Kosuge K;Uchida S;Hayashi H;Ohashi K, Hypertension, 2005, 28(3):223-227; Interaction between amlodipine and simvastatin in patients with hypercholesterolemia and hypertension 3-Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors are often prescribed in association with antihypertensive agents, including calcium antagonists. Simvastatin is an HMG-CoA reductase inhibitor that is metabolized by the cytochrome P450 (CYP) 3A4. The calcium antagonist amlodipine is also metabolized by CYP3A4. The purpose of this study was to investigate drug interactions between amlodipine and simvastatin. Eight patients with hypercholesterolemia and hypertension were enrolled. They were given 4 weeks of oral simvastatin (5 mg/day), followed by 4 weeks of oral amlodipine (5 mg/day) co-administered with simvastatin (5 mg/day). Combined treatment with simvastatin and amlodipine increased the peak concentration (C<inf>max</inf>) of HMG-CoA reductase inhibitors from 9.6±3.7 ng/ml to 13.7±4.7 ng/ml (p<0.05) and the area under the concentration-time curve (AUC) from 34.3±16.5 ng h/ml to 43.9±16.6 ng h/ml (p<0.05) without affecting the cholesterol-lowering effect of simvastatin. This study is the first to determine prospectively the pharmacokinetic and pharmacodynamic interaction between amlodipine and simvastatin Masica AL;Azie NE;Brater DC;Hall SD;Jones DR, Br J Clin Pharmacol, 2000, 50:273-276; Intravenous diltiazem and CYP3A-mediated metabolism AIMS: To study whether intravenous diltiazem, a calcium channel blocker commonly prescribed for hypertension and stable angina, is an inhibitor of the CYP3A enzymes by using oral lovastatin, an HMG Co-A reductase inhibitor, as a substrate. METHODS: Ten healthy volunteers were studied in a randomized two-way crossover design. The two arms were 1) administration of a 20 mg dosage of lovastatin orally and 2) administration of a 20 mg dosage of lovastatin orally 1 h after an intravenous loading dosage and constant infusion of diltiazem. Blood samples were collected up to 25 h in order to quantify lovastatin and diltiazem concentrations in the separated serum. Lovastatin and diltiazem concentrations were quantified by GC-MS and h.p.l.c., respectively. RESULTS: Intravenous diltiazem did not significantly affect the oral AUC, Cmax, t(1/2), or tmax of lovastatin. CONCLUSIONS: These data suggest that the interaction of lovastatin with diltiazem does not occur systemically and is primarily a first-pass effect. Thus, drug interactions with diltiazem may become evident when a patient is moved from intravenous to oral dosing Prasad PP;Stypinski D;Vyas KH;Gonasun L, Clin Drug Investig , 2004, a; Lack of interaction between modified-release fluvastatin and amlodipine in healthy subjects Objective: To investigate the potential for a pharmacokinetic interaction between fluvastatin modified-release 80mg tablet (LescolR XL; fluvastatin XL) and amlodipine 5mg tablet (NorvascR) following multiple once-a-day doses for 2 weeks. Design: This was a single-centre, six-sequence, three-period, randomised, cross-over design study. Fluvastatin XL 80mg tablet and amlodipine 5mg tablet were administered once a day for 2 weeks either alone or in combination. Fluvastatin and amlodipine serum concentration profiles were characterised on day 14 for each treatment. The pharmacokinetic interaction between the two drugs was evaluated based on the p-values and 90% confidence intervals (CIs) for log-transformed highest observed concentration (C <inf>max</inf>), area under the plasma concentra-tion-time curve calculated by the linear trapezoidal method up to 24 hours (AUC<inf>24</inf>), and apparent oral clearance at steady state (CL/F), using a single entity as the reference treatment and the combination as the test treatment. Adverse events (AEs), safety laboratory tests and physical examinations were evaluated for safety. Study participants: Twenty-four healthy subjects were enrolled and 19 completed the study. The safety analysis was based on data from all 24 subjects who received at least one dose of a treatment, while the pharmacokinetic analysis was based on data from the 19 subjects who completed all treatments. Results: The coadministration of fluvastatin XL and amlodipine resulted in no significant changes in the steady-state AUC (469 vs 454 < mu >g asterisk inside a circle sign h/L), C<inf>max</inf> (96 vs 89 < mu >g/L), and CL/F (197 vs 232 L/h) of fluvastatin when compared with fluvastatin XL alone. The p-values for these comparisons were between 0.172 and 0.238, and the 90% CIs for the geometric means were within 78% and 139%. A similar comparison for amlodipine showed no significant difference in the steady-state AUC (132 vs 140 < mu >g asterisk inside a circle sign h/L), C<inf>max</inf> (7.1 vs 7.5 < mu >g/L) and CL/F (41 vs 40 L/h) of amlodipine. The p-values for these comparisons were between 0.309 and 0.353, and the 90% CIs for the geometric means were within 90% and 111%. The majority of the AEs were mild in severity. There were no clinically relevant changes in clinical laboratory results, physical examinations or vital sign parameters. Conclusion: There were no significant differences in the steady-state pharmacokinetics of fluvastatin or amlodipine when they were administered together and the small differences observed were not clinically relevant. Therefore, no dose adjustment of either drug is necessary when fluvastatin and amlodipine are coadministered Park CG;Lee H;Choi JW;Lee SJ;Kim SH;Lim HE, Int J Clin Pharmacol Ther, 2010, a, 48:497-503; Non-concurrent dosing attenuates the pharmacokinetic interaction between amlodipine and simvastatin OBJECTIVES: To explore if non-concurrent amlodipine dosing results in less drug interaction, the pharmacokinetic profiles, safety and efficacy endpoints were assessed following repeated doses of simvastatin, co-administered concurrently or non-concurrently with amlodipine in patients with coexisting hypertension and hyperlipidemia. METHODS: Seventeen patients randomly received daily doses of 20 mg simvastatin and 5 mg amlodipine for 6 weeks, either with both drugs at 7:00 PM (concurrent) or with simvastatin at 7:00 PM followed by amlodipine at 11:00 PM (non-concurrent). The maximum plasma concentration (Cmax) and the area under the concentration-time curve up to the last quantifiable concentration (AUClast) were estimated at steady state. Lipid profiles and blood pressure values were also compared between the concurrent and non-concurrent groups. RESULTS: The Cmax and AUClast and of simvastatin acid in the non-concurrent amlodipine dosing group were 63.2% and 66.0%, respectively, of the values obtained in the concurrent group (1.2 +/- 1.0 vs. 1.9 +/- 0.9 ng/ml and 10.3 +/- 8.3 vs. 15.6 +/- 7.5 h ng/ml, respectively, mean +/- standard deviation). Changes from baseline in lipid profile and blood pressure were comparable between the groups. CONCLUSIONS: Non-concurrent dosing may be a useful and safe therapeutic option for patients who require two or more drugs administered concomitantly, but who are likely to develop unwanted drug interactions Watanabe H;Kosuge K;Nishio S;Yamada H;Uchida S;Satoh H;Hayashi H;Ishizaki T;Ohashi K, Life Sci, 2004, 76:281-292; Pharmacokinetic and pharmacodynamic interactions between simvastatin and diltiazem in patients with hypercholesterolemia and hypertension Pharmacokinetic and pharmacodynamic interactions between simvastatin, a 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor, and diltiazem, a calcium antagonist, were investigated in 7 male and 4 female patients with hypercholesterolemia and hypertension. The patients were given, for one in a three consecutive 4-week periods, oral simvastatin (5 mg/day), oral simvastatin (5 mg/day) combined with diltiazem (90 mg/day), and then oral diltiazem (90 mg/day), respectively. The area under the plasma concentration versus time curve up to 6 hours post-dose (AUC<inf>0-6h</inf>) and maximum plasma concentrations (Cmax) of the drugs, serum lipid profiles, blood pressures and liver functions were assessed on the last day of each of the three 4-week periods. After the combined treatment period, Cmax of HMG-CoA reductase inhibitor was elevated from 7.8 ± 2.6 ng/ml to 15.4 ± 7.9 ng/ml (P < 0.01) and AUC<inf>0-6h</inf> from 21.7 ± 4.9 ng< middle-dot >hr/ml to 43.3 ± 23.4 ng< middle-dot >hr/ml (P < 0.01), while Cmax of diltiazem was decreased from 74.2 ± 36.4 ng/ml to 58.6 ± 18.9 ng/ml (P < 0.05) and its AUC<inf>0-6h</inf> from 365 ± 153 ng< middle-dot >hr/ml to 287 ± 113 ng< middle-dot >hr/ml (P < 0.01). Compared to simvastatin monotherapy, combined treatment further reduced LDL-cholesterol levels by 9%, from 129 ± 16 mg/dl to 119 ± 17 mg/dl (P < 0.05). No adverse events were observed throughout the study. These apparent pharmacokinetic interactions, namely the increase of HMG-CoA reductase inhibitor concentration by diltiazem and the decrease of diltiazem concentration by simvastatin, enhance the cholesterol-lowering effects of simvastatin during combined treatment. < copyright > 2004 Elsevier Inc. All rights reserved Zhou LX;Finley DK;Hassell AE;Holtzman JL, J Pharmacol Exp Ther, 1995, 273:121-127; Pharmacokinetic interaction between isradipine and lovastatin in normal, female and male volunteers We have examined the pharmacokinetic interaction between isradipine and lovastatin in six male and six female, healthy, normotensive, human subjects after a single dose and after treatment for 5 days. The isradipine plasma concentrations were determined by a radioimmunoassay and the lovastatin serum concentrations by gas chromatography-mass spectrometry (GC/MS) and by the inhibition of the 3-hydroxy-3- methylglutaric-coenzyme reductase activity. We found that the apparent serum concentrations of lovastatin were 4- to 6-fold higher in the reductase-inhibition assay than the GC/MS assay, suggesting that the bulk of the reductase inhibition is due to active metabolites. The peak and the time-to-peak concentrations were unaffected by the treatments, either after the first dose or after continued administration. In male subjects, after repeated doses of isradipine, the lovastatin area under the time-concentration curves (AUCs) decreased by 40% as determined by the GC/MS assay (P < .001) and 20% as determined by the reductase- inhibition assay (P < .0022). In the female subjects, isradipine treatment decreased the lovastatin AUCs as determined by the GC/MS assay, but this was not statistically significant due to a high variance. Furthermore, in the female subjects, isradipine had no effect on the lovastatin AUCs as determined by the reductase-inhibition assay. Because the lovastatin peak and the time-to-peak concentrations were unaffected by isradipine treatment, the decreased lovastatin AUCs were probably not due to altered intestinal absorption. More likely, because lovastatin has a high hepatic clearance, the decreased AUCs seen after isradipine treatment could be due to increases in the clearance of lovastatin secondary to increased hepatic blood flow Choi D;Chung J;Choi J, Eur J Clin Pharmacol, 2010;285-290; Pharmacokinetic interaction between oral lovastatin and verapamil in healthy subjects: Role of P-glycoprotein inhibition by lovastatin Background Lovastatin is an inhibitor of P-glycoprotein (Pgp) and is metabolized by the cytochrome P450 (CYP) 3A4 isoenzyme. Verapamil is a substrate of both P-gp and CYP3A4. It is therefore likely that lovastatin can alter the absorption and metabolism of verapamil. Methods The pharmacokinetic parameters of verapamil and one of its metabolites, norverapamil, were compared in 14 healthy male Korean volunteers (age range 22-28 years) who had been administered verapamil (60 mg) orally in the presence or absence of oral lovastatin (20 mg). The design of the experiment was a standard 2x2 crossover model in random order. Results The pharmacokinetic parameters of verapamil were significantly altered by the co-administration of lovastatin compared to the control. The area under the plasma concentration-time curve(AUC<sub>0-</sub>) and the peak plasma concentration of verapamil were significantly increased by 62.8 and 32.1%, respectively. Consequently, the relative bioavailability of verapamil was also significantly increased (by 76.5%). The (AUC<sub>0-</sub>) of norverapamil and the terminal half-life of verapamil did not significantly changed with lovastatin coadministration. The metabolite-parent ratio was significantly reduced (29.2%) in the presence of lovastatin.Conclusion Lovastatin increased the absorption of verapamil by inhibiting P-gp and inhibited the first-pass metabolism of verapamil by inhibiting CYP3A4 in the intestine and/or liver in humans. Springer-Verlag 2009 Gladding P;Pilmore H;Edwards C, Ann Intern Med, 2004, 140:W31; Potentially fatal interaction between diltiazem and statins To describe the interaction between diltiazem and simvastatin-atorvastatin and highlights its clinical significance Peces R;Pobes A, Nephron, 2001, a, 89:117-118; Rhabdomyolysis associated with concurrent use of simvastatin and diltiazem Rhabdomyolysis due to interaction of simvastatin with the calcium channel blocker mibefradil has been reported. However, to the best of our knowledge, no case of rhabdomyolyse has been reported in association with diltiazem. We present a patient treated with both simvastatin and diltiazem who developed severe myositis and rhabdomyolysis. Rowan C;Brinker AD;Nourjah P;Chang J;Mosholder A;Barrett JS;Avigan M, Pharmacoepidemiol Drug Saf, 2009, 18:301-309; Rhabdomyolysis reports show interaction between simvastatin and CYP3A4 inhibitors PURPOSE: To assess spontaneous reports of rhabdomyolysis associated with simvastatin (SV) and pravastatin (PV) for evidence of CYP3A4 interaction. Clinical trial results advocate cholesterol lowering in high-risk patients including diabetics and the elderly. Given the association between advancing age, metabolic, and cardiovascular disease, many patients are treated with concomitant medications upon statin initiation. Although statins are generally safe, minor and severe adverse reactions arise, especially when given to patients taking concomitant medications that inhibit the statin clearance and lead to increased statin plasma concentration. METHODS: We conducted a comparative case series of rhabdomyolysis reports associated with SV and PV. Domestic spontaneous reports were obtained from the FDA's Adverse Event Reporting System (AERS). Drug utilization data were obtained from IMS HEALTH and the National Ambulatory Medical Care Survey (NAMCS). Adverse event reporting rates (AER) and ratios (AERR) of rhabdomyolysis associated with SV and PV-with and without stratification by CYP3A4 inhibitor concomitancy were determined. RESULTS: Stratification by CYP3A4 inhibitor concomitancy did not change the rhabdomyolysis AER for PV with or without a CYP3A4 inhibitor (2.4 cases and 3.1 cases per 10 million Rx, respectively). However, stratification of SV reports with or without a concomitant CYP3A4 inhibitor resulted in a rhabdomyolysis AER (38.4 and 6.0 cases per 10 million Rx, respectively). The corresponding AERR with or without a CYP3A4 inhibitor were 0.77 for PV and 6.43 for SV. CONCLUSIONS: Spontaneous adverse event reports provide evidence of increased risk for rhabdomyolysis based on interaction between SV and selected CYP3A4 inhibitors Lewin JJ;Nappi JM;Taylor MH, Ann Pharmacother, 2002, b, 36:1546-1549; Rhabdomyolysis with concurrent atorvastatin and diltiazem OBJECTIVE: To report a case of rhabdomyolysis and acute hepatitis associated with the coadministration of atorvastatin and diltiazem. CASE SUMMARY: A 60-year-old African American man with a significant past medical history presented to the emergency department with acute renal failure secondary to rhabdomyolysis. In addition, liver enzymes were elevated to greater than 3 times normal. The only change in medication was the initiation of diltiazem 3 weeks earlier for atrial fibrillation to a complicated medication regimen that included atorvastatin. DISCUSSION: Rhabdomyolysis has been reported in patients receiving hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase inhibitors when coadministered with agents that may inhibit their metabolism. Atorvastatin is the most potent of this class of agents currently available and is commonly used in the treatment of hyperlipidemia. Rhabdomyolysis resulting from the drug interaction between diltiazem and other HMG-CoA reductase inhibitors has been described in the literature. However, no report has specifically associated this adverse event with atorvastatin and diltiazem. We describe a patient with a complex medication regimen who was admitted for rhabdomyolysis and accompanying acute renal failure, along with acute hepatitis, thought to be secondary to a drug interaction between atorvastatin and diltiazem. CONCLUSIONS: While optimizing the patient's lipid profile should be the primary factor in choosing one statin over another, the potential for drug interactions requires close attention. All patients beginning HMG-CoA reductase inhibitor therapy should be counseled regarding the signs and symptoms of muscle injury; particular attention should be paid to those patients who are taking medications that may interact Kanathur N;Mathai MG;Byrd RP;Fields CL;Roy TM, Tenn Med, 2001, a, 94:339-341; Simvastatin-diltiazem drug interaction resulting in rhabdomyolysis and hepatitis Simvastatin, a hydroxymethyl glutarate coenzyme A (HMG-CoA) reductase inhibitor, is a commonly used cholesterol lowering agent. The long-term safety profile of simvastatin, established over ten-years of clinical use, is excellent. Both rhabdomyolysis and hepatitis, however, are recognized toxic effects of this medication, and generally occur when the patients are taking more than 40 mg of simvastatin a day. Potent inhibitors of the cytochrome P450 3A4 (CYP3A4) enzyme increase the incidence of simvastatin toxicity. Calcium channel blockers are weak inhibitors of the CYP3A4 enzyme. Diltiazem is known to increase the serum concentration of simvastatin. Many patients who take both simvastatin and diltiazem require lower doses of simvastatin to achieve the recommended reduction in cholesterol. Since diltiazem is known to increase plasma levels of lovastatin, a similar phenomenon may occur with simvastatin. Our patient had been stable for three years on simvastatin therapy. His rhabdomyolysis and hepatitis coincided with the addition of diltiazem. This is the first report of the combined toxicities of rhabdomyolysis and hepatitis being induced by the addition of diltiazem to simvastatin therapy. This patient serves as a reminder to the clinician of the potential interaction of these two commonly used drugs Ziviani L;Da Ros L;Squassante L;Milleri S;Cugola M;Iavarone LE, Br J Clin Pharmacol, 2001, 51:147-152; The effects of lacidipine on the steady/state plasma concentrations of simvastatin in healthy subjects AIMS: Lacidipine, a long acting 2, 4-dihydropyridine calcium channel antagonist is frequently administered with cholesterol lowering agents, particularly in elderly populations. The effects of lacidipine on the pharmacokinetics of simvastatin were investigated, since they share the CYP3A4 pathway for metabolism. METHODS: The study was an open, randomised, two-way crossover design, with at least 7 days washout. Eighteen healthy subjects received simvastatin, 40 mg once daily, alone and together with lacidipine, 4 mg once daily, for 8 days. The pharmacokinetics of simvastatin were studied on the eighth day. Analysis was made of total simvastatin acid concentrations (naive simvastatin acid plus that derived from alkaline hydrolysis of the lactone). RESULTS: Lacidipine increased the maximum concentration of simvastatin (Cmax) by approximately 70% (P=0.016) and the area under the plasma concentration-time curve AUC(0,24 h) by approximately 35% (P=0.001). The mean Cmax and AUC(0,24 h) of simvastatin (95% confidence interval) when given alone were 8.76 (6.72-11.41) ng ml(-1) and 60.36 (47.15-77.28) ng ml(-1) h. During treatment with lacidipine they were, respectively, 14.89 (10.77-20.58) ng ml(-1) and 80.96 (64.62-101.44) ng ml(-1) h. No significant differences were observed in either time to peak concentration (tmax was 1.0 h for simvastatin alone and 1.5 h for the combination) or in the half-life (t1/2,z was 8.5 h in both cases). The combination was safe and well tolerated. CONCLUSIONS: The observed increased exposure to simvastatin 40 mg following coadministration of lacidipine is unlikely to be of clinical relevance Azie NE;Brater DC;Becker PA;Jones DR;Hall SD, Clin Pharmacol Ther, 1998, 64:369-377; The interaction of diltiazem with lovastatin and pravastatin BACKGROUND: Lovastatin is oxidized by cytochrome P4503A to active metabolites but pravastatin is active alone and is not metabolized by cytochrome P450. Diltiazem, a substrate and a potent inhibitor of cytochrome P4503A enzymes, is commonly coadministered with cholesterol- lowering agents. METHODS: This was a balanced, randomized, open-label, 4-way crossover study in 10 healthy volunteers, with a 2-week washout period between the phases. Study arms were (1) administration of a single dose of 20 mg lovastatin, (2) administration of a single dose of 20 mg pravastatin, (3) administration of a single dose of lovastatin after administration of 120 mg diltiazem twice a day for 2 weeks, and (4) administration of a single dose of pravastatin after administration of 120 mg diltiazem twice a day for 2 weeks. RESULTS: Diltiazem significantly (P < .05) increased the oral area under the serum concentration-time curve (AUC) of lovastatin from 3607 +/- 1525 ng/ml/min (mean +/- SD) to 12886 +/- 6558 ng/ml/min and maximum serum concentration (Cmax) from 6 +/- 2 to 26 +/- 9 ng/ml but did not influence the elimination half-life. Diltiazem did not affect the oral AUC, Cmax, or half-life of pravastatin. The average steady-state serum concentrations of diltiazem were not significantly different between the lovastatin (130 +/- 58 ng/ml) and pravastatin (110 +/- 30 ng/ml) study arms. CONCLUSION: Diltiazem greatly increased the plasma concentration of lovastatin, but the magnitude of this effect was much greater than that predicted by the systemic serum concentration, suggesting that this interaction is a first-pass rather than a systemic event. The magnitude of this effect and the frequency of coadministration suggest that caution is necessary when administering diltiazem and lovastatin together. Further studies should explore whether this interaction abrogates the efficacy of lovastatin or enhances toxicity and whether it occurs with other cytochrome P4503A4- metabolized 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors, such as simvastatin, fluvastatin, and atorvastatin Mousa O;Brater DC;Sunblad KJ;Hall SD, Clin Pharmacol Ther, 2000, 67:267-274; The interaction of diltiazem with simvastatin BACKGROUND: Simvastatin is an inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase that is used as a cholesterol-lowering agent and is metabolized by cytochrome P450 3A (CYP3A) enzymes. Diltiazem is a substrate and an inhibitor of CYP3A enzymes and is commonly coadministered with cholesterol-lowering agents such as simvastatin. The objective of this study was to quantify the effect of diltiazem on the pharmacokinetics of simvastatin. METHOD: A fixed-order study was conducted in 10 healthy volunteers with a 2-week washout period between the phases. In one arm of the study, a single 20-mg dose of simvastatin was administered orally; the second arm entailed administration of a single 20-mg dose of simvastatin orally after 2 weeks of treatment with 120 mg diltiazem twice a day. RESULTS: Diltiazem significantly increased the mean peak serum concentration of simvastatin by 3.6-fold (P < .05) and simvastatin acid by 3.7-fold (P < .05). Diltiazem also significantly increased the area under the serum concentration-time curve of simvastatin 5-fold (P < .05) and the elimination half-life 2.3-fold (P < .05). There was no change in the time to peak concentration for simvastatin and simvastatin acid. CONCLUSION: Diltiazem coadministration resulted in a significant interaction with simvastatin, probably by inhibiting CYP3A-mediated metabolism. Concomitant use of diltiazem or other potent inhibitors of CYP3A with simvastatin should be avoided, or close clinical monitoring should be used SPC for Zanidip, Tomt indhold, 2007; Zanidip (lercanidipin), filmovertrukne tabletter
|
|
|
|
|
|
|